DOI:
10.1039/B208335C
(Paper)
J. Mater. Chem., 2003,
13, 143-149
One-step synthesis and characterization of ZrO2–WOx prepared by hydrothermal method at autogenous pressure
Received 27th August 2002, Accepted 1st November 2002
First published on 20th November 2002
Abstract
A series of ZrO2–WOx samples was hydrothermally prepared in an autoclave at autogenous pressure in a range of temperatures from 145 to 225 °C for 24 h. The materials were characterized by elemental analysis, X-ray diffraction, nitrogen physisorption and FT-IR spectroscopy of both structure and adsorbed pyridine. The Rietvelt refinement of the X-ray diffraction patterns was also performed. The formation of crystalline products was detected at synthesis temperatures as low as 145 °C. Pure zirconium oxihydroxide gel yielded a mixture of metastable t-ZrO2
(63%) and m-ZrO2
(37%) zirconia phases when treated at 145 °C. When tungsten (15 wt.% as WO3) was added to the synthesis mixture, only an amorphous phase of t-ZrO2 was detected. Both the crystallization of t-ZrO2 phase and the t-ZrO2 to m-ZrO2 phase transformation, caused by hydrothermal treatment, were retarded by the presence of tungsten. In this way a microcrystalline ZrO2–WOx of 309 m2 g−1 was prepared under hydrothermal conditions at 145 °C. The material becomes progressively more crystalline with increasing synthesis temperature. At 225 °C a well-crystallized material (79% of t-ZrO2 and 21% of m-ZrO2 phases) was obtained. Interestingly, the crystallite sizes of both phases were not too different, 15.6 and 13 nm for t-ZrO2 and m-ZrO2 respectively. In all the samples, crystalline phases associated with WOx species were not unambiguously identified by XRD. FT-IR spectroscopy of W-promoted zirconia showed a band at 943 cm−1 not present in non-tungsten-promoted ZrO2, which was attributed to the W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode of octahedrally coordinated tungsten species. Since the lattice parameters of the ZrO2 structure were not modified by the presence of tungsten, the WOx species must be highly dispersed and strongly bound to the ZrO2 surface. Post-synthesis calcination treatment at higher temperatures (560, 700 and 800 °C) brought about sintering of these dispersed tungsten species and the formation of a more bulky tungsten oxide species characterized by two FT-IR vibration bands at 970 and 1100 cm−1.
O stretching mode of octahedrally coordinated tungsten species. Since the lattice parameters of the ZrO2 structure were not modified by the presence of tungsten, the WOx species must be highly dispersed and strongly bound to the ZrO2 surface. Post-synthesis calcination treatment at higher temperatures (560, 700 and 800 °C) brought about sintering of these dispersed tungsten species and the formation of a more bulky tungsten oxide species characterized by two FT-IR vibration bands at 970 and 1100 cm−1.
1 Introduction
Because of its interesting physico-chemical properties, zirconium oxide (ZrO2) is a material with increasing use in catalysis.1 This material has a high melting point (2350 °C), low thermal conductivity and high corrosion resistance.2 From a chemical point of view, zirconia is an amphoteric material with both oxidizing and reducing properties.3 However, the strong acidic properties of zirconia, when promoted with sulfate ZrO2–SO42− or tungsten (ZrO2–WOx), have undoubtedly attracted the most attention over the past few years.4–7 ZrO2–WO3, with presumably a lower acidic strength but a higher stability, seems to be the most promising candidate to replace liquid acid catalysts and the halogen-promoted aluminas, which are subject to ever more severe environmental regulation. From current understanding of tungsten-doped zirconia there are two parameters that are important in the preparation of a solid with adapted catalytic properties: the W loading (10–15 wt.%) and the activation temperature (usually around 800 °C). Both calcination temperature and tungsten loading correspond to those in which tungsten species become active for the catalysis. However, Iglesia et al.8,9 have recently shown evidence that WOx active structures depend only on the WOx surface density and not on the WOx concentration or oxidation temperature independently. Another feature of this system is that zirconia must crystallize in the pure tetragonal phase for optimal catalytic properties. However, for ZrO2–WO3 as for ZrO2–SO42−, the appropriate treatment temperature will mainly depend on the synthesis method.7 Considering that ZrO2–WO3 samples require relatively high calcination temperatures (>800 °C), an important loss of pore volume and surface area is always observed in this type of material.The key to catalytic applications of tungsten-doped zirconia is the control of its textural and acidic properties. The synthesis parameters and the crystallization process are directly related to those properties. Different procedures for increasing the surface area of sulfate- or tungsten-doped zirconia have been explored. One of the most frequently used is the “sol–gel” method, which yields a very porous material if combined with supercritical drying.10 Ko et al.11–13 have reported the synthesis of ZrO2 doped with sulfate, tungsten and a mixture of both promoters by the sol–gel technique. They showed not only the gain in the textural properties but also in the catalytic performance of the materials. We have shown14,15 that ZrO2–SO42− prepared by sol–gel with in situ sulfation is about six times more active than that prepared by the classical two-step method. Santiesteban et al.16 combined the co-precipitation method with a digestion procedure to prepare a ZrO2–WOx which was three times more active than those obtained by impregnation or standard co-precipitation techniques. More recently, G. K. Chuah et al.17,18 reported an easy route to improve the surface area of zirconia by digesting the hydrous zirconia precursor before drying and calcination. The resulting zirconia had a surface area around 250 m2 g−1 after treatment at 500 °C. Through this route of preparation, the metastable tetragonal form of zirconia persisted even up to 1000 °C.
For catalysis applications a decrease in the activation temperature of ZrO2–WO3 would lead to a material with a higher surface area and pore volume. However, this temperature must be sufficient for the activation of both the metastable tetragonal ZrO2 phase and the tungsten oxide active phase. In the present context, the hydrothermal method may provide an alternative route for the ZrO2–WO3 synthesis. Among the advantages of this method are the following: a comparatively low processing temperature, an in situ metal oxide and growth promoter, the possibility of selective growth (only on active layers or phases), the possibility of batch processing, etc.
In this work we present a hydrothermal route for the synthesis of a crystalline zirconia with a well dispersed tungsten oxide species on its surface. A series of tungsten-doped zirconias was prepared by the hydrothermal method inside an autoclave at temperatures between 145–225 °C and autogenous pressure. The degree of zirconia crystallization as well as the type of tungsten oxide crystalline structure depends on the synthesis temperature.
2 Experimental
2.1 Synthesis of materials
A mixed zirconium–tungsten hydroxide was precipitated from aqueous solutions of zirconyl nitrate hydrate [ZrO(NO3)2·6H2O, Aldrich 99.99%] 0.16 M and ammonium metatungstate hydrate [(NH4)6W12O39
× H2O, Aldrich 99%
+80 mesh] 0.007 M. The coprecipitation was carried out at constant pH (9.5–10) using an aqueous solution (14% vol) of NH4OH (J. T. Baker, 28 wt.%) as the precipitating agent. The ammonium metatungstate solution was placed in a 2000 ml beaker and the zirconyl nitrate and ammonium hydroxide solutions were added slowly to this solution. A constant and vigorous agitation was maintained during the coprecipitation procedure. In order to keep the pH of coprecipitation constant, the rate of addition of both solutions was adjusted. The obtained slurry was placed into a 1000 ml autoclave reactor, and then it was carefully sealed. The hydrothermal synthesis was carried out under autogenous pressure at 145, 165 and 225 °C over 24 h. The resulting material was then filtered and dried at 120 °C for 18 h. As reference, a ZrO2–WO3 sample was prepared by precipitation-digestion-calcination method. The following procedure was used. A mixed zirconium–tungsten hydroxide was precipitated from aqueous solutions of zirconyl oxychloride hydrate [ZrOCl2·6 H2O, Aldrich 99.99%] 0.16 M and ammonium metatungstate hydrate [(NH4)6W12O39
× H2O, Aldrich 99%
+80 mesh] 0.007 M. The resulting slurry was refluxed at 100 °C for 24 h, and then cooled to room temperature, filtered and dried at 120 °C for 18 h. The samples are coded as follows: WZr-CH-T1(T2) for samples co-precipitated and hydrothermally treated, where T1 is the hydrothermal synthesis temperature and T2 is the post-synthesis calcination temperature, WZr-CD-T2 for the sample prepared by coprecipitation-digestion method. The absence of W means a non-tungsten-promoted zirconia.2.2 Characterization
The tungsten content of samples was determined by atomic absorption spectroscopy on a Perkin-Elmer 5000 spectrophotometer. The X-ray diffraction (XRD) patterns of the samples were recorded at room temperature with CuKα radiation in a Bruker Advanced D-8 diffractometer having a theta–theta configuration and a secondary beam monochromator. Diffraction intensity was measured in the 2θ range between 15 and 110° with a 2θ step of 0.02 ° with 8 s per point. Crystalline structures were refined by the Rietvelt technique using FULLPROF V3.5d codes.19 Peak profiles were modelled with a pseudo-Voigt function20 containing average crystallite size as one of its characteristic parameters.21 Standard deviations of the Rietvelt refined parameters are given in parenthesis on Table 1. These values are not the estimation of the probable error in the analysis as a whole, but only of the minimum possible errors based on their normal distribution.22 For the Rietvelt refinement of the crystalline structure, the tetragonal zirconia unit cell symmetry was described with the p42/nmc space group. The monoclinic zirconia was modelled with the unit cell having the P21/c space group. The WO3 was modelled using an orthorhombic unit cell with the Pmb symmetry of space group.
Table 1 Crystallite size and phase composition of the synthesized and annealed WZr-CH sample. As a referenced the WZr-CD-800 sample is shown
| Samples | Annealing temperature/°C | Tetragonal ZrO2/ | Monoclinic ZrO2/ |
|---|
| wt.% | nm | wt.% | nm |
|---|
| Zr-CH-145 | 110 | 63(2) | 10.5(1) | 37.0(3) | 12.5(2) |
| WZr-CH-165 | 110 | 34.0(6), 49(6) | 12.9(4), 0.7(1) | 16(2) | 6.3(5) |
| WZr-CH-225 | 110 | 30(3), 48(4) | 17.5(8), 14.5(3) | 21.5(5) | 11.5(4) |
| WZr-CH-165 | 560 | 71(4) | 13.6(2) | 28(2) | 8.1(3) |
| WZr-CH-165 | 700 | 75.3(3) | 14.2(1) | 24(1) | 9.8(2) |
| WZr-CH-165 | 800 | 74.2(2) | 18.1(2) | 26(2) | 11.6(3) |
| WZr-CD | 800 | 59(2) | 15.7(2) | 34(2) | 16.1(5) |
BET surface areas and pore size distributions were obtained by nitrogen physisorption in an ASAP-2000 analyzer from Micromeritics.
The solids were characterized by FT-IR spectroscopy using a 170 SX Nicolet spectrometer. Self-supported wafers (≈14 mg) were mounted in a glass cell equipped with KBr windows, which permitted following the changes of the spectrum with thermal treatments. Before the IR spectrum measurement, the samples were degassed at 500 °C under a dried airflow of 60 ml min−1 for 1 h. For the acidity measurements, the sample was evacuated to 10−1 Pa, then maintained under a flow (2.14 l h−1) of nitrogen saturated with pyridine for 15 min, outgassed 1 h at room temperature, then heated up to the desired temperature using a linear ramp. The procedure to calculate the concentration of Brønsted and Lewis acid sites is described in ref. 23.
3 Results and discussion
3.1 X-Ray diffraction
3.1.1 Effect of synthesis temperature. The X-ray diffraction patterns of WZr-CH-145, WZr-CH-165, and WZr-CH-225 samples (using zirconyl nitrate hydrate as starting material) are shown on Fig. 1. For reference, the spectrum of the non-tungsten-promoted zirconia (Zr-CH-145 sample) is also shown. The crystalline phases composition of samples and their mean crystallite size derived from the Rietvelt refined parameters are presented in Table 1. Hydrothermal aging of hydrous amorphous pure zirconia gel at 145 °C yields a mixture of metastable t-ZrO2
(63%) and m-ZrO2
(37%) phases. It is clear that without a high temperature treatment a highly crystalline ZrO2 phase can be obtained. It is also worth noting that both crystalline phases presented similar crystallite sizes: 10.5 nm and 12.5 nm for the t-ZrO2 and for m-ZrO2 respectively (Table 1). It seems that both crystalline phases were already independently formed from the start of the synthesis. On the other hand, if one applies the concept of the crystallite size effect24 to explain the stabilization of the metastable t-ZrO2 phase at low temperature and its subsequent transformation to m-ZrO2 with temperature, the metastable t-ZrO2 crystalline phase would be being stabilized with a critical size at about 10 nm, approximately half of that reported by Garvie24 under non-hydrostatic stress. Thus, t-ZrO2 microcrystals annealed under hydrothermal conditions are more readily transformed to the monoclinic phase with only light grinding. Mitsuhashi et al.25 proposed that the strain generated at domain boundaries, and not the crystallite size effect, is responsible for the existence of metastable t-ZrO2. However, as Garvie also reported,26 the critical crystallite size at any given temperature is strongly dependent on any hydrostatic and/or non-hydrostatic stress present. |
| | Fig. 1 X-Ray diffraction patterns of ZrO2–WOx samples synthesized at different hydrothermal treatment conditions. (a) Zr-CH-145, (b) WZr-CH-145, (c) WZr-CH-165, (d) WZr-CH-225. Upper tick marks correspond to tetragonal ZrO2 phase and the lower ones to the monoclinic ZrO2 phase. | |
The tungsten presence strongly influences the development of the crystalline phases of zirconia. At the lowest hydrothermal crystallization temperature, the material was completely amorphous as measured by X-rays (WZ-CH-145 sample, Fig. 1b). As in the case of tungsten- or sulfate-doped zirconia prepared by non-hydrothermal methods, the incorporation of tungsten delays the transition of zirconia from the amorphous to the metastable t-ZrO2 phase.27–29 When temperature increased from 145 °C to 165 °C a decrease of the amorphous material, with the concomitant increase in intensity of diffraction lines ascribed to t-ZrO2, was observed. The growth of the metastable t-ZrO2 phase was favored over the m-ZrO2 phase. On WZr-CH-165 sample (Fig. 1c) only a 16% of m-ZrO2 phase was quantified. The rest of the spectrum was fitted using two metastable tetragonal zirconia phases. 34% of t-ZrO2 presented a crystallite size of 12.9 nm and the other portion (49%) was microcrystalline. Interestingly, the m-ZrO2 phase presented a smaller crystal size (6.3 nm) than that of the metastable tetragonal zirconia phase (12.9 nm). At this temperature a tungsten oxide phase was not detected clearly by X-ray diffraction. At 225 °C (WZr-CH-225 sample, Fig. 1d) practically all the remaining microcrystalline material crystallized into the metastable t-ZrO2 phase. Since the crystal size of the metastable t-ZrO2 phase remained practically constant, increasing the temperature favored the crystallization of the remaining amorphous material into the metastable t-ZrO2. Only a small increase of the m-ZrO2 phase was observed (from 16 to 21 wt.%); moreover the crystal size remained below that showed by the metastable t-ZrO2 phase. With respect to the crystallization of a tungsten oxide phase, only a broad signal in the region of the orthorhombic phase of crystalline WO3
(2θ
= 23.1, 23.6, and 24.4) was detected. Since the lattice parameters of zirconium oxide cell (Table 2) were not practically modified by the presence of tungsten oxide, these results suggest a good dispersion of tungsten oxide phase on the surface of the ZrO2 support.
Table 2 Lattice parameters of the synthesized and annealed WZr-CH sample. As a reference the WZr-CD-800 is shown
| Sample | Tetragonal | Monoclinic |
|---|
| a/nm | c/nm | a/nm | b/nm | c/nm |
|---|
| WZr-CH-165 | 0.35993(9) | 0.5203(2) | 0.5189(9) | 0.5004(9) | 0.5413(9) |
| WZr-CH-225(T1) | 0.35825(9) | 0.5197(2) | 0.5127(3) | 0.5175(3) | 0.5329(3) |
| WZr-CH-225(T2) | 0.35983(8) | 0.5237(2) | — | — | — |
| WZr-CH-165-560 | 0.35920(4) | 0.52005(7) | 0.5140(3) | 0.5178(4) | 0.5315(4) |
| WZr-CH-165-700 | 0.35938(2) | 0.52008(5) | 0.5133(2) | 0.5179(2) | 0.5331(2) |
| WZr-CH-165-800 | 0.35945(2) | 0.51956(4) | 0.5130(2) | 0.5192(2) | 0.5331(2) |
| WZr-CD-800 | 0.35940(2) | 0.51823(5) | 0.5140(1) | 0.5186(2) | 0.5338(1) |
3.1.2 Effect of post-synthesis calcination. In order to identify the structure of tungsten species (not detected by XRD at the synthesis temperature) present on hydrothermally synthesized ZrO2–WOx samples, the WZr-CH-165 sample was calcined in a flow of dry air at 560, 700 and 800 °C. Fig. 2 shows the XRD patterns for the WZr-CH-165 sample over the range of activation temperatures. As a reference the diffractogram of the WZr-CD-800 sample, prepared by the coprecipitation-digestion method and calcined at 800 °C, is also shown. As can be seen in Fig. 2, the orthorhombic phase of WO3 was not clearly detected even when a post-synthesis treatment at 800 °C was carried out on the WZr-CH-165 sample. At this calcination temperature the lines assigned to the orthorhombic WO3 phase in WZr-CD sample were already identified. In a recent paper of Shon and Park,30 for a 13-WO3–ZrO2 sample prepared by adding an aqueous solution of ammonium metatungstate to Zr(OH)4 the triclinic phase of WO3 appeared at 400 °C. In the square zoom of Fig. 2, a comparison (only on the orthorhombic WO3 region) between the WZr-CH-165-800 and WZr-CD-800 samples showed that it is not possible to assign the peak observed in this region on the WZr-CH-165-800 sample to some tungsten oxide phase. On the other hand, from Fig. 2 it can be observed that once the WZr-CH-165 sample has been completely crystallized (Fig. 2a) the ZrO2 phase composition of the sample did not change with the post-synthesis calcinations at 560, 700 and 800 °C. Over the entire temperature range the t-ZrO2 phase remained in the 71–75 wt.% range. As indicated in Table 1, only an increase in the crystallite size (from 13.6 to 18.1 nm) of the t-ZrO2 phase was observed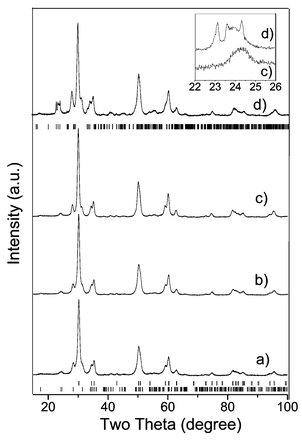 |
| | Fig. 2 X-Ray diffraction patterns of WZr-CH-165 sample annealed at (a) 560 °C, (b) 700 °C, (c) 800 °C, and (d) WZr-CD annealed at 800 °C. Upper tick marks correspond to orthorhombic WO3 phase, middle tick marks correspond to tetragonal ZrO2 phase and the lower ones to the monoclinic ZrO2 phase. | |
3.2 Main physico-chemical characteristics
The effect of preparation conditions on the textural properties of ZrO2–WOx was analyzed. On the WZr-CH-165 sample, the role of the post-synthesis activation temperature was also examined. The main physico-chemical characteristics of all the samples are shown in Table 3. As a reference, the data obtained from WZr-CD-800 sample were also included. Interestingly, the hydrothermal synthesis of non-tungsten-promoted zirconia (Zr-CH-145 sample) yielded a well-crystallized material, as discussed earlier, with 136 m2 g−1 of surface area. This area was higher than those reported as very high for ZrO2–WOx samples prepared by the sol–gel method.17 The increasing surface area was attributed to the smaller crystallites dimensions produced by the hydrothermal method: 10.5 nm and 12.5 nm for t-ZrO2 and m-ZrO2 phases respectively. We have previously reported this same effect for Fe-Zn-Al-O mixed oxides.31 It is worth noting that the presence of tungsten increases considerably the BET surface area of zirconia. For the microcrystalline tungsten-doped zirconia (WZr-CH-145 sample) a surface area of 308 m2 g−1 was measured. WZr-CH-165 and WZr-CH-225 samples had surface areas of 169 m2 g−1 and 107 m2 g−1 respectively. As reported in the literature, the increase in surface area is attributed to the inhibition of sintering process by the presence of the dopant.16,27–29 After calcination at 700 °C the WZr-CH-165 sample retained a surface area of 94 m2 g−1. Considering that this material was crystallized at low temperature, the activation at higher temperatures is not critical for surface area decreasing. By contrast, as will be discussed later, this post-synthesis thermal pretreatment has a positive effect on the acidity of these materials. After calcination at 700 °C of WZr-CH-165 sample, the decreasing of the surface area was explained by both the amorphous or microcrystalline t-ZrO2 to crystalline t-ZrO2 transformation and the increasing crystallite size of the m-ZrO2 phase as shown in Table 1.
Table 3 Textural properties and tungsten density of the synthesized and annealed WZr-CH sample. As a reference the WZr-CD-800 sample is shown
| Sample | Annealing temperature/°C | Surface area/m2 g−1 | Porous volume/cm3 g−1 | Mean porous size/Å | W/atoms nm−2 |
|---|
| |
|---|
| Zr-CH-145 | 110 | 136 | 0.212 | 62.62 | 0 |
| WZr-CH-145 | 110 | 308 | 0.400 | 51.90 | 1.23 |
| WZr-CH-165 | 110 | 169 | 0.275 | 65.19 | 2.25 |
| WZr-CH-225 | 110 | 107 | 0.194 | 72.68 | 3.56 |
| WZr-CH-165 | 700 | 94 | 0.204 | 86.63 | 4.06 |
| WZr-CH-165 | 800 | 60 | 0.239 | 159.73 | 6.55 |
| WZr-CD | 800 | 66 | 0.694 | 419.1 | 7.44 |
3.3 Pore structure
The nitrogen adsorption/desorption isotherms of zirconia–tungsten samples synthesized at different temperatures are shown in Fig. 3. For all the samples the isotherms are nearly identical in shape, being essentially type IV according to BDDT classification.32 The hysteresis loop is type A according to Boer's classification.33 This type of isotherm is characteristic of systems with open or closed pores of variable radius. They have also been associated with ink-bottle pores where the entrance of the pores is narrower than the body. From Fig. 3 it is clear that the volume adsorbed at low pressure decreases with increasing synthesis temperature. As was observed by X-Ray diffraction, increasing the synthesis temperature brings about the growth of zirconia crystallites with the concomitant loss of microporous area. Therefore, the higher surface area of zirconia synthesized at low temperature is mainly associated with the presence of microcrystalline material. The relationship between the BET surface area and the crystallite size is shown in Fig. 4. This figure shows the BJH desorption average pore diameter of all the samples. It is clear that by increasing the synthesis temperature a larger crystallite size of ZrO2 is favored. The larger the particles are, the more the pore size distribution is shifted toward larger pore sizes and consequently the average pore diameter is increased, as shown in Table 3. However, as evidenced by the adsorption/desorption isotherms, this increase is not too large, from about 5.2 to 7.2 nm. From Fig. 4 it can also be seen that the pore size distribution falls in a small range. As a consequence of the synthesis temperature increasing, the growth of smaller t-ZrO2 particles was favored over bigger ones. Thus, if the crystal size of t-ZrO2 remains small the transformation of the metastable t-ZrO2 phase to m-ZrO2 phase will be minimized. On the other hand, although the samples calcined at 800 °C (WZr-CH-165-800 and WZr-CD-800 samples) had practically the same surface area, the mean pore diameter distribution (Fig. 4b) showed a unimodal distribution for the sample prepared by the hydrothermal method and a bimodal distribution for the sample prepared by the classical precipitation-calcination method (WZr-CD-800 sample). This means that the WZr-CH-165-800 sample has a more homogeneous crystallite size distribution. |
| | Fig. 3 Nitrogen adsorption/desorption isotherms of samples synthesized at different temperature (a) WZr-CH-145, (b) WZr-CH-165, (c) WZr-CH-225, (d) WZr-CH-165-800, (e) WZr-CD-800. | |
 |
| | Fig. 4 Pore diameter distribution of samples synthesized at different temperature (a) WZr-CH samples, (b) WZr-CH-165-800 sample. WZr-CD-800 sample is included as reference. | |
3.4 FT-IR study
3.4.1 FT-IR of ZrO2–WOx samples. In order to elucidate the nature of the surface tungsten species on ZrO2, FT-IR spectroscopy was used. The vibrational spectra of Zr-CH-145 and WZr-CH-165 samples, registered at room temperature and 400 °C, are presented in Fig. 5A and B respectively. The spectra of the WZr-CH-165 sample annealed at 560, 700 and 800 °C are also included in this same Figure. The FT-IR spectrum of the 15WZr-CD sample is also added for reference. In contrast to others studies29,30 FT-IR analysis of dry and wet surfaces did not show any significant differences. Because of the samples prepared by the hydrothermal method showed a well-defined crystalline structure at low temperature, practically no difference between the spectra registered at room temperature and 400 °C was expected. The FT-IR spectrum of the prepared WZr-CH-165 sample, showed three bands with maxima centered at about 757, 943 and 1043 cm−1. For a 10wt.% W–ZrO2 sample prepared by precipitation and sol–gel methods Boyse and Ko34 reported a band at 750 cm−1, which was assigned to a W–O vibration of surface tungstate species. They established that this band possibly comes from both crystalline and non-crystalline species. In the present study the band at 757 cm−1, clearly visible in the WZr-CD-800 sample, can be assigned to this type of surface tungsten species. However, the fact that this band is observed in the Zr-145 sample (non tungsten-promoted sample) suggests that it can be also assigned to a Zr–O vibration. The bands at 943 and 1043 cm−1 also correspond to W–O vibrations of surface tungstate species in strong interaction with the ZrO2 surface. However, these species would be different from those correlated to the 750 cm−1 band. Boyse and Ko assigned the infrared bands in the range of 970 to 990 cm−1 to solvated polytungstate species. In the present study, the shift to lower wavenumbers (943 cm−1) means that a stronger zirconia–tungsten interaction exists. It is interesting to note that when the FT-IR analysis was made at 400 °C (Fig. 5B), practically all the IR bands remained at the same wavenumber. Considering that ZrO2 lattice parameters were not perturbed by the presence of tungsten, one can assign the band at 943 cm−1 to the WO stretching mode of the highly dispersed WO3 crystallites. These isolated WOx species would be stabilized through multiple Zr–O–W bonds between the octahedral WO6 and zirconia surface. These tungsten species correspond to WOx isolated groups reported by Iglesia et al.8,9 for a tungsten surface density below 4 W nm−2. In the present study the WZr-CH-165 sample had a tungsten surface density of 2.25 W nm−2. From Fig. 6Aa or Fig. 6Ba it can be seen that the band at 943 cm−1 is not present on non tungsten-promoted ZrO2. When the WZr-CH-165 sample was calcined at higher temperatures (560, 700 and 800 °C), in a post-synthesis pretreatment, this band shifted to 970 cm−1. At the same time as the activation temperature increased a new band at 1100 cm−1 was detected. This later band is clearly observed with high intensity on the WZr-CD-800 sample. Because on this same sample the XRD spectrum showed the orthorhombic WO3 crystallites, this unambiguously indicates the connection between the 1100 cm−1 band and the orthorhombic WO3 phase. These results are in accord with those reported by Iglesia et al.8 and Boyse and Ko.34 They reported the emergence of a crystalline WO3 phase with increasing activation temperature. X-ray diffraction patterns of the WZr-CH-165 sample, annealed at 560, 700 and 800 °C (Fig. 2b, 2c and 2d respectively) did not clearly show the octahedrally coordinated WO3 species. However, by analogy with the same IR bands showed by the ZW-CD-800 sample in which the orthorhombic phase of WO3 was clearly detected by XRD, one can assign the band at 1100 cm−1 to the presence of some fraction of an orthorhombic WO3 phase, and the band at 970 cm−1 to a polytungstate structure (with tungsten octahedrally coordinated) as was proposed by Iglesia et al.9 This band appears at lower wavenumber than those reported in the literature at 1000–1020 cm−1.29,30 For 20wt.% WO3/ZrO2 calcined at 750 °C, Shon and Park30 observed a single band at 1003–1012 cm−1, which was assigned to the symmetrical WO stretching mode of the tungsten oxide complex bonded to the ZrO2 surface. As reported by Boyse and Ko34 and Shon and Park, as the evacuation temperature increases, the WO stretching mode shifts to higher wavenumbers (from 1003 to 1012 cm−1). Moreover, Boyse and Ko have shown that the most active sample in butane isomerization corresponded to the sample with only one peak present at 1008 cm−1, which was associated with surface WOx species. This same temperature-dependent behavior was also observed by Ramis et al.35 on tungsta–titania catalysts. These authors identified the active surface species on the dried WO3–TiO sample as coordinately unsaturated “monooxo wolframyls” having a very short WO bond. In the present work, considering the much stronger zirconia–tungsten interaction, this band is shifted to a lower wavenumber. The band at 1100 cm−1, clearly observed in the ZW-CD-800 sample and scarcely observed in hydrothermal samples, has not been reported in the literature. This band could be associated with a well-crystallized WO3 phase. The high wavenumber of this band indicates a high bond order of the structure associated to it.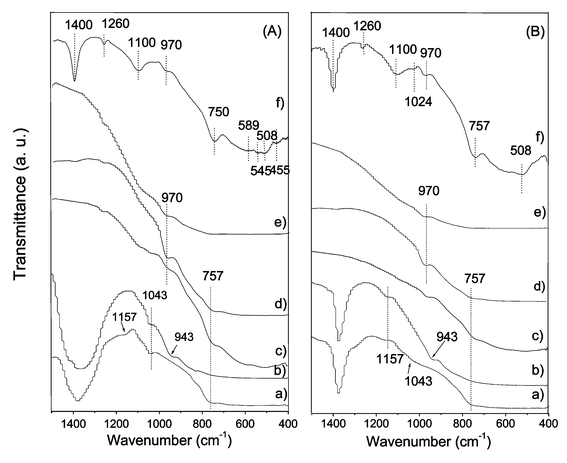 |
| | Fig. 5 FT-IR spectra of the ZrO2–WOx samples. (A) analyzed at 25 °C, (B) analyzed at 400 °C. (a) Zr-CH-145, (b) WZr-CH-165, (c) WZr-CH-165-560, (d) WZr-CH-165-700, (e) WZr-CH-165-800, and (f) WZr-CD-800. | |
 |
| | Fig. 6 FT-IR spectra of pyridine adsorbed on ZrO2–WOx samples. (A) after evacuation at 50 °C, (B) after evacuation at 400 °C. (a) Zr-CH-145, (b) WZr-CH-165, (c) WZr-CH-165-700, (d) WZr-CH-165-800, and (e) WZr-CD-800. | |
3.4.2 Acidity properties of ZrO2–WOx samples. The acidic properties of zirconia–tungsten were studied to identify the relationship between the nature of tungsten surface species and the acidity of the materials. The FT-IR spectra of adsorbed pyridine were used to determine both the type of acid sites (Brønsted or Lewis) and the relative strengths of these acid sites. The FT-IR spectra of adsorbed pyridine on Zr-CH-145 and WZr-CH-165 samples, evacuated at 50 and 400 °C are shown in Fig. 6A and B respectively. The spectra of the WZr-CH-165 sample, annealed at 700 and 800 °C are also showed. As a reference, the spectrum of WZr-CD-800 sample was included. For the as-prepared samples (Fig. 6Aa and b), the existence of Lewis acid sites was clearly evident by the peaks at 1609 and 1489 cm−1. After evacuation of pyridine at 400 °C these high intensity bands were still present. On these spectra a broad and pronounced band, which extended from about 1500 to 1580 cm−1, is also clearly evident. If one considers that this band can envelop the vibration band at 1540 cm−1, associated with Brønsted acidity, then these two samples would possess both types of acidity. With respect to this band Figueras et al.36 have observed that pyridine adsorbed in Lewis acid sites can react with neighbors OH to produce α-pyridone, with IR vibrational bands in the 1500–1600 cm−1 region. Then, although these authors observed the formation of α-pyridone after heating at 400 °C and we at 50 °C, this broad band can be attributed to this species. On the other hand, the evolution of the acidity with respect to the activation temperature of the synthesized samples is shown in the spectra of Fig. 6Ac and d corresponding to WZr-CH-165 sample calcined at 700 and 800 °C respectively. After calcination at 700 °C the band at 1540 cm−1, associated with Brønsted acidity is now clearly defined. The subsequent calcination at 800 °C brings about a decrease of the bands associated with Lewis acidity with an increase of the bands at 1639 and 1540 cm−1 corresponding to Brønsted acidity. Then, it is clear that the post-synthesis calcination temperature strongly influences the acidity of the WZr-CH-165 sample. On the surface of the synthesized sample only Lewis acidity was detected. When the sample was subjected to calcination Brønsted acidity was favored. The analysis of the spectrum of WZr-CD-800 sample showed that at low temperature its profile is similar to that of the WZr-CH-165 sample calcined at 800 °C. However, after evacuation at 400 °C the WZr-CD-800 sample does not retain pyridine; whereas for the WZr-165 sample calcined at 800 °C the bands associated with Lewis and Brønsted acidity are still visible. This illustrates that although all the samples possessed Lewis and Brønsted acidity, the samples prepared by the hydrothermal method adsorb pyridine irreversibly at 400 °C. Conclusions
ZrO2–WOx crystalline samples were prepared by hydrothermal treatments of amorphous mixed zirconium–tungsten hydroxide gels at temperatures as low as 165 °C. Zirconia phase composition and crystallite size are functions of the synthesis temperature. Compared with a classical precipitation-calcination method, the hydrothermal method yielded a ZrO2–WOx sample with a stronger zirconium–tungsten interaction. A crystalline WOx phase was not detected by XRD at any synthesis temperature. The post-synthesis treatment at higher temperatures (700 and 800 °C) decreases the zirconia surface area with the concomitant increasing of the WOx surface density. Even at these high temperatures, a crystalline WOx phase was not clearly observed. On the synthesized samples Lewis acidity was mainly measured by FT-IR spectroscopy of adsorbed pyridine. When WOx surface density increased, the Brønsted acidity was preferentially favored.Acknowledgement
This work was financially supported by IMP projects D.00065 and D.01234References
- Catal. Today, 1994, 20, 1 Search PubMed.
- P. D. L. Mercera, J. G. Van Ommen, E. B. M. Doesburg, A. J. Burggraaf and J. R. H. Ross, Appl. Catal., 1991, 71, 363 CrossRef CAS.
- K. Tanabe, Mater. Chem. Phys., 1985, 13, 347 CrossRef CAS.
- K. Arata, Adv. Catal., 1990, 37, 165 Search PubMed.
- T. Yamagushi, Appl. Catal., 1990, 61, 1 CrossRef CAS.
- M. Hino and K. Arata, J. Chem. Soc. Chem. Commun., 1988, 1259 RSC.
- H. Armendáriz, B. Coq, D. Tichit, R. Dutartre and F. Figueras, J. Catal., 1998, 173, 345 CrossRef CAS.
- D. G. Barton, M. Shtein, R. D. Wilson, S. T. Soled and E. Iglesia, J. Phys. Chem. B, 1999, 103, 630 CrossRef CAS.
- C. D. Baertsch, S. L. Soled and E. Iglesia, J. Phys. Chem. B, 2001, 105, 1320 CrossRef CAS.
- C. J. Brinker and G. W Scherer, in Sol-Gel Science. The Physics and Chemistry of the Sol-Gel Processing, Academic Press, Boston, 1990 Search PubMed.
- D. A. Ward and E. Ko, J. Catal., 1994, 150, 18 CrossRef CAS.
- D. A. Ward and E. Ko, J. Catal., 1995, 157, 321 CrossRef CAS.
- D. A. Ward and E. Ko, J. Catal., 1998, 179, 100 CrossRef.
- D. Tichit, B. Coq, H. Armendáriz and F. Figueras, Catal. Lett., 1996, 38, 109 Search PubMed.
- H. Armendáriz, C. Sanchez Sierra, F. Figueras, B. Coq, C. Mirodatos, F. Lefebvre and D. Tichit, J. Catal., 1997, 171, 86.
- J. G. Santiesteban, J. C. Vartuli, S. Han, R. D. Bastian and C. D. Chang, J. Catal., 1997, 168, 431 CrossRef CAS.
- G. K. Chuah, S. Jaenicke and B. K. Pong, J. Catal., 1998, 175, 80 CrossRef CAS.
- G. K. Chuah, Catal. Today, 1999, 49, 131 CrossRef CAS.
- J. Rodriguez-Carbajal, Laboratoire Leon Brillouin (CEA-CNRS), France.
- P. Thompson, D. E. Cox and J. B. Hasting, J. Appl. Crystallogr., 1987, 20, 79 CrossRef CAS.
- R. A. Young and P. Arch. Desai, Nauki Mat., 1989, 10, 71 Search PubMed.
- E. Prince, J. Appl. Crystallogr., 1978, 14, 157 CrossRef.
- M. Pérez, H. Armendáriz, J. A. Toledo, A. Vázquez, J. Navarrete, A. Montoya and A. García, J. Mol. Catal. A: Chem., 1999, 149, 169 CrossRef CAS.
- R. C. Garvie, J. Phys. Chem., 1965, 69(4), 1238 CrossRef CAS.
- T. Mitsuhashi, M. Ichihara and U. Tatsuke, J. Am. Cer. Soc., 1973, 57(2), 97 Search PubMed.
- R. C. Garvie, J. Phys. Chem., 1978, 82(2), 218 CrossRef CAS.
- R. Srinivasan, D. Taulbee and B. H. Davis, Catal. Lett., 1991, 9, 1 Search PubMed.
- M. S. Scurrel, Appl. Catal., 1987, 34, 109 CrossRef CAS.
- T. Yamagushi and K. Tanabe, Mater. Chem. Phys., 1986, 16, 67 CrossRef.
- J. R. Shon and M. Y. Park, Langmuir, 1998, 14, 6140 CrossRef.
- J. A. Toledo, M. A. Valenzuela, P. Bosch, H. Armendáriz, A. Montoya, N. Nava and A. Vázquez, Appl. Catal. A: General, 2000, 198, 235 CrossRef CAS.
- S. Brunauer, L. S. Deming, W. S. Deming and E. Teller, J. Am. Chem. Soc., 1940, 62, 1723 CrossRef CAS.
- S. Lowell, in Introduction To Powder Surface Area, John Wiley and Sons, New York, 1979 Search PubMed.
- R. A. Boyse and E. I. Ko, J. Catal., 1997, 171, 191 CrossRef CAS.
- G. Ramis, G. Busca, C. Cristiani, L. Lietti, P. Forzatti and F. Bregani, Langmuir, 1992, 8, 1744 CrossRef CAS.
- S. Bodoardo, F. Figueras and E. Garrone, J. Catal., 1994, 147, 223 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2003 |
Click here to see how this site uses Cookies. View our privacy policy here.