DOI:
10.1039/B205258J
(Paper)
J. Mater. Chem., 2003,
13, 21-26
Nonlinear optical studies of fullerene–arylethyne hybrids†
Received
31st May 2002
, Accepted 5th November 2002
First published on 25th November 2002
Abstract
The fullerene–arylethyne hybrids 2 and 3 were synthesized in order to investigate their NLO activities. The β values for 2 and 3 were obtained by hyper Rayleigh scattering measurements. The arylethynyl fullerenes 3a–f exhibit β values in the range 20–361 × 10−30 esu. Interestingly, the β values for 3a,b are higher than those for 3c–f; a good correlation was obtained between the Hammett σp values of the para substituents of 3 and their β values. The 1H-NMR and E1/2 data indicate that the fullerene moieties of 3 attract electrons from the arylethynyl groups in the ground state. The β values for 2a–c are higher than those for 3a–f; the para-carborane–fullerene dyad 2c exhibits an exceptionally high β value (1189 × 10−30 esu). The electronic characteristics of the carbon and boron clusters are discussed.
Introduction
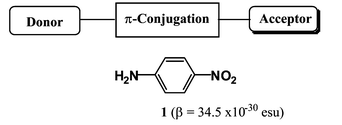
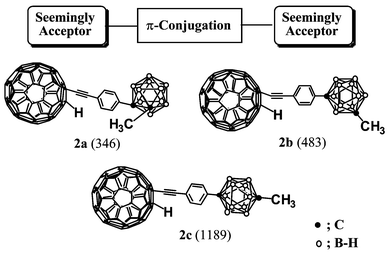
Recent years have witnessed a dramatic growth in multidisciplinary research activity involving materials that exhibit quadratic nonlinear optical (NLO) behavior, which have attracted attention owing to their direct application in the development of efficient optical telecommunication networks without electrical-to-optical, and vice versa, signal conversion.1 Such applications require thermally robust materials with highly nonlinear optical responses (first hyperpolarizability, β). The use of organic materials may offer significant advantages over conventional inorganic crystals such as LiNbO3.2 In the past decade, considerable efforts have been focused on the development of materials with large molecular hyperpolarizabilities. Typical organic NLO compounds include donor (D) and acceptor (A) moieties connected via a π-conjugated linker. Until now, most efforts to obtain better hyperpolarizabilities have been directed towards finding both the right combination of D and A and the right length of conjugated bridge between D and A3 [for example, 4-nitroaniline (1); Scheme 1]. On the other hand, we recently reported that combinations of seemingly attractive carboranes and seemingly attractive fullerene linked through an arylethynyl π-system (2a–c; Scheme 2) give unexpectedly high β values, with the para-carborane derivative 2c showing the highest β value (1189 × 10−30 esu).4
 |
| | Scheme 1 | |
 |
| | Scheme 2 | |
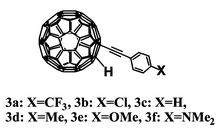
It has been known for many years that closo-carboranes are electron-deficient boron clusters with highly polarizable σ-aromatic character5 and that fullerene is an electron-attracting carbon cluster having highly delocalizable π-electrons.6 Carboranes (A) containing electron-donor (D) moieties have been synthesized, but their β values were found to be moderate or low.7 Nevertheless, the fullerene–carborane hybrid dyads 2a–c, which are seemingly acceptor–acceptor combinations, exhibit unexpectedly high NLO activities; β values of 346, 483, and 1189 × 10−30 esu for 2a–c, respectively.4 Accordingly, we decided to investigate systematically whether the fullerene moieties of 2 really act as acceptors in NLO activities, as is believed and often mentioned in the literature.8,9 We synthesized the arylethynyl fullerenes 3, in which appropriate electron-donating and electron-withdrawing groups (X) were attached to the para position of the phenyl ring (Scheme 3). There was a good correlation between the β values of 3 and the Hammett σp values of the X substituents. In this article, we report full details of this interesting result, together with the role of carborane in NLO activities, which explains why compounds 2 exhibit rather high β values.
 |
| | Scheme 3 | |
Experimental
NMR, UV-vis, cyclic voltammetry (CV) and hyper Rayleigh scattering (HRS) spectra are provided in the ESI.
Instrumentation
Fluorescence and UV-vis absorption spectra were measured with a Hitachi F-4500 spectrofluorophotometer and a Hitachi U-3000 UV-vis spectrometer, respectively. The HRS measurements were carried out by using a q-switched Nd∶YAG laser (spectron LS-412; fwhm 6 ns) as an incident light beam. The fundamental light intensity [γ
= 1064 nm, I(ω)] was varied by rotating a half-wave plate positioned between two polarizers. The HRS signal, I(2ω) was detected by a photomultiplier (Philips model XP 2020Q) as a function of I(ω). Both incident fundamental light and HRS light signals were fed into two gated integrators (Stanford Research Systems model SR250) and output signals from the gated integrators were A–D converted and analyzed on a personal computer. NMR spectra were recorded at frequencies of 300 MHz for 1H and 150 MHz for 13C, from CDCl3–CS2 solutions. FAB mass spectra were obtained using a JEOL JMS-HX110 spectrometer using m-nitrobenzyl alcohol matrices.
Materials
C60
(99.5%) was purchased from MER Corp. The phenylacetylene derivatives 4a–f were prepared according to the published procedure.10 The fullerene–carborane dyads 2a–c4 and phenylethynylfullerene 3c11 were synthesized by the reported procedures.
General procedure for the synthesis of 3
Under a scrupulously dry Ar atmosphere, a 1.5 M n-hexane solution of n-BuLi (0.2 ml, 0.3 mmol) was added to a THF solution (1.5 ml) of the appropriate phenylacetylene derivative 4
(0.3 mmol) at 0 °C. The solution became yellowish orange and was left at room temperature for 15 min. A THF solution (6.0 ml) of C60
(72 mg, 0.1 mmol) was sonicated for 30 min. at 30 °C under a dry Ar atmosphere. The solution of lithiated 4 was added slowly to the C60 solution. The mixture turned dark green and was stirred vigorously for 15 min. After addition of CF3COOH (2 ml), the solvents were evaporated. Silica gel column chromatography (with n-hexane, Et2O, then CH2Cl2 as eluents) of the resulting brown–black crude products gave compounds 3 in the yields indicated in Table 1.
Table 1 Yields of 3 from the reaction of fullerene with 4
| Compound |
X |
Yield (%) |
|
Ref. 11.
|
|
3a
|
CF3 |
20 |
|
3b
|
Cl |
6 |
|
3c
|
H |
39a |
|
3d
|
Me |
40 |
|
3e
|
OMe |
44 |
|
3f
|
NMe2 |
47 |
1-{4-(2-Methyl-1,2-dicarba-closo-dodecaboran-1-yl)phenyl}ethynyl-1,2-dihydrofullerene-60 (2a).
4
1H-NMR: δ 7.82 (d, J
= 8.7 Hz, 2H), 7.76 (d, J
= 8.7 Hz, 2H), 7.11 (s, 1H), 1.78 (s, 3H). 13C-NMR: δ 151.12, 150.66, 146.52, 146.42, 146.37, 146.22, 145.73, 145.69, 145.48, 145.45, 145.44, 145.34, 144.67, 144.44, 143.21, 142.99, 142.62, 142.58, 142.05, 141.97, 141.75, 141.66, 141.61, 140.39, 140.37, 135.92, 135.21, 132.38, 131.33, 131.21, 130.89, 125.07, 100.39, 95.13, 82.02, 61.63, 55.32, 29.79. MS (FAB+)
m/z: 978 (M+); 817.0 [M+
−
(1-Me-1,2-C2B10H11)]; 721 (C60H); 720 (C60); 257 (1-CCPh,2-Me-1,2-C2B10H10); 234 (1-Me,2-Ph,1,2-C2B10H10); 158 (1-Me-1,2-C2B10H10).
1-{4-(7-Methyl-1,7-dicarba-closo-dodecaboran-1-yl)phenyl}ethynyl-1,2-dihydrofullerene-60 (2b).
4
1H-NMR: δ 7.68 (d, J
= 8.4 Hz, 2H), 7.51 (d, J
= 8.4 Hz, 2H), 7.10 (s, 1H), 1.82 (s, 3H). 13C-NMR: δ 151.18, 150.84, 147.50, 147.22, 146.49, 146.31, 146.28, 146.13, 146.11, 145.67, 145.45, 145.40, 145.34, 145.25, 144.58, 144.38, 143.12, 142.91, 142.52, 142.49, 141.99, 141.95, 141.90, 141.72, 141.59, 141.53, 140.28, 135.92, 135.74, 135.10, 131.88, 128.00, 122.97, 93.78, 82.59, 61.69, 55.06, 24.56. MS (FAB+)
m/z: 977 (M+
− H); 817.0 [M+
−
(1-Me-1,7-C2B10H11)]; 744 [M+
−
(1-Me,7-Ph-1,7-C2B10H10)]; 721 (C60H); 720 (C60); 258 (1-CCPh,7-Me-1,7-C2B10H10); 234 (1-Me,7-Ph,1,7-C2B10H10); 158 (1-Me-1,7-C2B10H10).
1-{4-(12-Methyl-1,12-dicarba-closo-dodecaboran-1-yl)phenyl}ethynyl-1,2-dihydrofullerene-60 (2c).
4
1H-NMR: δ 7.54 (d, J
= 8.5 Hz, 2H), 7.25 (d, J
= 8.5 Hz, 2H), 7.05 (s, 1H), 1.23 (s, 3H). 13C-NMR: δ 151.28, 150.97, 147.53, 147.25, 146.53, 146.34, 146.31, 146.15, 145.71, 145.61, 145.51, 145.41, 145.36, 145.27, 144.61, 144.42, 143.14, 142.95, 142.54, 142.50, 142.01, 141.93, 141.76, 141.61, 141.55, 140.30, 140.28, 136.74, 135.96, 135.12, 131.66, 127.43, 122.55, 93.47, 82.72, 61.72, 55.01, 25.57. MS (FAB+)
m/z: 977 (M+
− H); 817.0 [M+
−
(1-Me-1,12-C2B10H11)]; 744 [M+
−
(1-Me,12-Ph-1,12-C2B10H10)]; 721 (C60H); 720 (C60); 258 (1-CCPh,12-Me-1,12-C2B10H10); 234 (1-Me,12-Ph-1,12-C2B10H10); 158 (1-Me-1,12-C2B10H10).
1-(4-Trifluoromethylphenyl)ethynyl-1,2-dihydrofullerene-60 (3a).
1H-NMR: δ 7.92 (d, J
= 9 Hz, 2H), 7.74 (d, J
= 9 Hz, 2H), 7.13 (s, 1H). 13C-NMR: δ 151.16, 150.70, 147.59, 147.31, 146.54, 146.41, 146.36, 146.21, 145.74, 145.70, 145.50, 145.48, 145.43, 145.33, 144.66, 144.44, 143.20, 142.98, 142.61, 142.57, 142.05, 141.97, 141.76, 141.66, 141.61, 140.38, 135.94, 135.24, 132.39, 130.67 (q, JC–F
= 33 Hz), 126.25, 125.43 (m), 123.67 (q, JC–F
= 271 Hz), 94.61, 82.17, 61.64, 54.99. MS (FAB+)
m/z: 891 (M+
+ H), 745 (M+
− CF3C6H4), 720 (C60).
1-(4-Chlorophenyl)ethynyl-1,2-dihydrofullerene-60 (3b).
1H-NMR: δ 7.74 (d, J
= 8.7 Hz, 2H), 7.45 (d, J
= 8.7 Hz, 2H), 7.11 (s, 1H). 13C-NMR: δ 151.41, 151.12, 147.66, 147.38, 146.64, 146.45, 146.42, 146.26, 145.83, 145.71, 145.63, 145.51, 145.48, 145.39, 144.72, 144.52, 143.24, 142.65, 142.61, 142.13, 142.08, 142.02, 141.86, 141.72, 141.65, 140.41, 140.39, 136.05, 135.23, 135.20, 133.37, 128.93, 120.93, 93.22, 82.50, 61.76. MS (FAB+)
m/z: 859 (M+
+ H), 745 (M+
− ClC6H4), 720 (C60).
1-Phenylethynyl-1,2-dihydrofullerene-60 (3c).
12
1H-NMR: δ 7.77 (m, 2H), 7.45 (m, 3H), 7.13(s, 1H). 13C-NMR: δ 151.51, 151.22, 147.49, 147.21, 146.63, 146.30, 146.27, 146.12, 145.70, 145.60, 145.55, 145.41, 145.33, 145.25, 144.61, 144.42, 143.13, 142.52, 142.49, 142.06, 141.97, 141.93, 141.81, 141.62, 141.54, 140.30, 140.27, 135.99, 135.08, 132.06, 128.91, 128.55, 122.47, 92.24, 84.05, 61.87, 55.03. MS (FAB+)
m/z: 822 (M+), 720 (C60)
1-(4-Methylphenyl)ethynyl-1,2-dihydrofullerene-60 (3d).
1H-NMR: δ 7.69 (d, J
= 8 Hz, 2H), 7.27 (d, J
= 8 Hz, 2H), 7.12 (s, 1H), 2.45 (s, 3H). 13C-NMR: δ 151.47, 151.38, 147.50, 147.23, 146.57, 146.29, 146.12, 146.11, 145.72, 145.60, 145.56, 145.39, 145.33, 145.25, 144.60, 144.43, 143.11, 142.92, 142.50, 142.47, 141.95, 141.92, 141.81, 141.61, 141.52, 140.29, 140.23, 138.94, 136.04, 135.05, 132.03, 129.23, 119.38, 91.62, 83.90, 61.88, 55.09, 21.62. MS (FAB+)
m/z: 837 (M+
+ H), 744 (M+
− MeC6H4
− H), 720 (C60).
1-(4-Methoxyphenyl)ethynyl-1,2-dihydrofullerene-60 (3e).
1H-NMR: δ 7.73 (d, J
= 9 Hz, 2H), 7.12 (s, 1H), 6.98 (d, J
= 9 Hz, 2H), 3.89 (s, 3H). 13C-NMR: δ 151.59, 151.58, 147.57, 147.30, 146.63, 146.35, 146.18, 146.16, 145.78, 145.67, 145.60, 145.43, 145.38, 145.31, 144.65, 144.49, 143.16, 142.98, 142.55, 142.52, 142.09, 141.97, 141.88, 141.66, 141.57, 140.34, 140.27, 136.10, 135.06, 133.56, 114.46, 114.12, 91.00, 83.74, 61.94, 55.17, 55.12. MS (FAB+)
m/z: 853 (M+
+ H), 744 (M+
− MeOC6H4
− H), 720 (C60).
1-(4-N,N-Dimethylaminophenyl)ethynyl-1,2-dihydrofullerene-60 (3f).
1H-NMR: δ 7.66 (d, J
= 9 Hz, 2H), 7.12 (s, 1H), 6.75 (d, J
= 9 Hz, 2H), 3.06 (s, 6H). 13C-NMR: δ 152.14, 151.95, 150.47, 147.65, 147.38, 146.76, 146.41, 146.25, 146.23, 145.91, 145.89, 145.62, 145.48, 145.45, 145.38, 144.74, 144.61, 143.22, 142.60, 142.58, 142.19, 142.08, 142.04, 141.74, 141.63, 140.41, 140.28, 136.28, 135.09, 133.28, 111.87, 108.99, 90.28, 84.86, 62.13, 55.46, 40.17. MS (FAB+)
m/z: 866 (M+
+ H), 744 (M+
− MeOC6H4
− H), 720 (C60).
Results and discussion
Syntheses of the arylethynyl fullerenes 3
The arylethynyl fullerenes 3 were prepared in one step from the corresponding aryl acetylenes 4, which were synthesized by the Sonogashira coupling protocol; aryl iodides or bromides were treated with trimethylsilylacetylene under Pd(PPh3)4–CuI-catalyzed conditions.10 Treatment of 4a–f with n-BuLi, followed by addition of C60 and subsequent treatment with CF3COOH, gave 3a–f in moderate to low yields11
(Table 1). The synthesis of compounds 2 was carried out in a similar manner to that described previously;4 the carborane-substituted phenylacetylenes were synthesized and the coupling reaction between them and fullerene gave the corresponding carborane–fullerene dyads 2. The arylethynyl fullerenes 3c–f, having electron-donating groups at the para position of the phenyl ring, were obtained in higher yields than 3a,b, which have electron-withdrawing para groups. The lithiated arylethynes of 4a,b underwent facile polymerization, in comparison with those of 4c–f, leading to low yields of 3a,b. The structural assignments of 3a–f were carried out by analytical and spectroscopic techniques (i.e.1H-NMR, 13C-NMR, UV-vis and FAB MS) and full details are given in the ESI. Representative data from the UV-vis and 1H-NMR spectra of 3 and 4 are summarized in Table 2. The 1H-NMR spectra of 3 show a proton on the fullerene skeleton around δ 7.1, which is in good agreement with the chemical shifts reported for protons of related fullerene derivatives.11 Additionally, the protons on the benzene rings of 3 appear at slightly lower magnetic fields, compared to those of the corresponding compounds 4, indicating that in the ground state, the fullerene moieties of 3 attract electrons from the benzene ring.
| Compound |
X |
λ
max/nm (log ε) |
1H-NMR |
| C60Ha |
C6H4b |
|
Protons on benzene ring.
Proton on fullerene cage.
Ref. 12
|
|
3a
|
CF3 |
258 (5.29), 328 (4.76), 431 (3.70) |
7.29, 7.74 |
7.13 |
|
4a
|
— |
7.57 |
— |
|
3b
|
Cl |
258 (5.23), 328 (4.67), 432 (3.52) |
7.74, 7.45 |
7.11 |
|
4b
|
— |
7.39, 7.25 |
— |
|
3c
|
H |
258 (5.09), 328 (4.53),432 (3.41)c |
7.77, 7.45 |
7.13 |
|
4c
|
— |
7.45, 7.28 |
— |
|
3d
|
Me |
258 (5.40), 328 (4.87), 433 (3.62) |
7.69, 7.27 |
7.12 |
|
4d
|
— |
7.37, 7.11 |
— |
|
3e
|
OMe |
258 (5.12), 328 (4.56), 430 (3.52) |
7.73, 6.98 |
7.12 |
|
4e
|
— |
7.41, 6.82 |
— |
|
3f
|
NMe2 |
258 (5.07), 295 (4.88), 430 (3.75) |
7.66, 6.75 |
7.12 |
|
4f
|
— |
7.36, 6.61 |
— |
SHG activities of 3 and correlation between their β values and the Hammett σp values of the X substituents
In order to measure hyperpolarizability values with hyper Rayleigh scattering, it is essential that there are no absorptions or emissions around 532 nm, since the resonance and fluorescence around 532 nm enhance the hyperpolarizabilities of 3, which makes it difficult to obtain accurate β values. The UV-vis absorption spectra of 3a–e in CHCl3 show two large absorption bands at 258 and 328 nm, which arise from fullerene moieties (Table 2).11 In the case of 3f, a strong band appears at 295 nm instead of 328 nm. The NMe2 group is well known as an auxochrome and, in general, the absorption of phenyl rings with NMe2 substituents appears around 300 nm. In this case, the absorption of the phenylacetylene moiety of 3f appears at 295 nm and an enhanced auxochrome effect due to the NMe2 group is observed. The UV-vis spectrum of 3f is different from the spectra of 3b–e because the strong absorption of the (dimethylaminophenyl)acetylene moiety obscures the absorption of the fullerene moiety at 328 nm. The spectrum of 3a shows a weak absorption at 310 nm. This could be due to the phenylacetylene moiety. In the case of 3b–e, however, similar absorptions around 300 nm are not observed, perhaps because the strong absorptions of fullerene at 258 and 328 nm obscure any peaks around 300 nm. Furthermore, very weak absorption bands are observed around 430–433 nm (log ε 3.41–3.75), which are presumably due to a forbidden transition; absorption bands around 430 nm are typical of 1,2-dihydrofullerenes.12 No absorptions are observed at wavelengths greater than 450 nm for CHCl3 solutions of 3a–f, although C60 itself and 3c in cyclohexane exhibit very weak absorption bands between 470–700 nm with log ε 2.3–3.2. Excitation at 258 and 328 nm produced fluorescence spectra, which show broad band emissions in the range 350–390 nm. The emission spectra are attributed to Raman scattering by CHCl3. Fullerene fluorescence is usually observed around 700 nm and is, in general, measured in cyclohexane.13 However, in the present case using CHCl3 as solvent, fluorescence spectra around 700 nm were not observed. We attempted to dissolve 3a,b and 3d–f in cyclohexane, THF and CH3CN, but were unsuccessful. In conclusion, CHCl3 solutions of 3
(10−4 M) did not show significant spectroscopic absorption and emission around the double frequency value 532 nm, allowing us to carry out the measurements necessary to deduce the β coefficients.
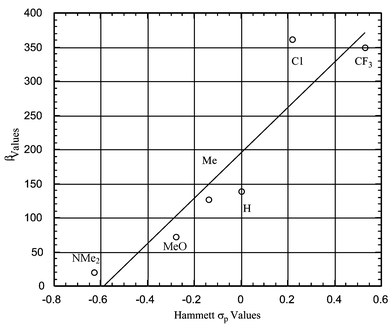
HRS measurements were performed to determine the molecular nonlinear optical properties of the dyads. All the measurements were performed at a fundamental wavelength of 1064 nm (see Experimental). The β values for 3a–f are summarized in Table 3. It should be noted that 3a,b, which are fullerene–acceptor systems, exhibit much higher β values than 3c–f, fullerene–donor systems. As expected, the β value for fullerene itself was zero under the same experimental conditions, due to its centrosymmetric structure. Fig. 1 shows a correlation between the β values for 3 and the Hammett σp values of the X substituents.
 |
| | Fig. 1 Plot of Hammett σpversusβ for 3. | |
Table 3 Hammett σp and β values for 3
| Compound (X) |
σ
p
|
β/10−30 esu |
β
0/10−30 esu |
|
3a
(CF3) |
0.53 |
350 |
196 |
|
3b
(Cl) |
0.22 |
361 |
ca. 220 |
|
3c
(H) |
0 |
139 |
ca. 83 |
|
3d
(Me) |
−0.14 |
127 |
ca. 76 |
|
3e
(OMe) |
−0.28 |
72 |
ca. 43 |
|
3f
(NMe2) |
−0.63 |
20 |
15 |
A good correlation between the β values for 3 and the Hammett σp values of the X substituents is observed; higher σp values lead to higher β values, indicating that fullerene acts as an electron-donating group in the excited state. As mentioned above, fullerene acts as an electron-withdrawing group in the ground state, and this is an accepted fact in fullerene chemistry.6,12 As discussed below, the NLO activities depend on the electronic characteristics of the excited process.
The calculation of β0 values is essential for determining the energy levels of empty orbitals, which exert the strongest influence on the single harmonic generation (SHG) activity. Considering the correlation between the Hammett σp values and the β values for 3, it is reasonable to conclude that the orbitals of the phenylacetylene moieties around 300 nm exert the strongest influence on the SHG activities. The β0 values for 3a and 3f, which were calculated according to the two-level model, are shown in Table 3. The exact β0 values for 3b–e could not be determined because the absorptions of phenylacetylene moieties were not observed, as mentioned above. Accordingly, the β0 values for 3b–e were calculated based upon the assumption that the UV absorptions would appear at 300 nm, similarly to those of 3a and 3f.
Electrochemical studies of 2 and 3c,d
In order to clarify the role of the fullerene moieties of 3 in the NLO activities and the role of the carboranes of 2 in the high hyperpolarizabilities, the electrochemical behavior of 2a–c and 3c,d, which are essentially reflections of the LUMO levels of the fullerene moieties, were studied by cyclic voltammetry in o-dichlorobenzene at room temperature. The redox properties of 3a,b were also examined, but we could not obtain satisfactory data, perhaps because the carbon–halogen bonds of 3a,b are cleaved under the electrochemical conditions employed. The cyclic voltammograms of 2a and C60 are shown in Fig. 2.11a Compounds 2a–c and 3c,d show similar CV curves, and reversible reduction waves corresponding to the reduction of fullerene moieties were observed. The redox potentials of 2a–c, 3c,d and C60 are summarized in Table 4, in which the first reductive potential (E1red), the first oxidative potential (E1ox) and E1/2 [1/2(E1red
+
E1ox)] are shown for simplicity, although three quasi-reversible reduction–oxidation peaks were observed, as shown in Fig. 2 for 2a. The one-electron reduction of uncharged molecules of 2 and 3c,d
(E1red), and the one-electron oxidation of 2 and 3c,d
(E1ox) are more important than further reductions and the oxidations of the charged molecules in the discussion of the electrochemical properties of 2 and 3c,d. The E1red values for 2a–c and 3d are more negative than that for fullerene itself, and the E1ox values for 2a–c and 3c,d are between −770 and −550 mV. In terms of the electronic properties of 2 and 3c,d, both the E1red and E1ox values are important because they give the information about the electron-transfer ability of the substrates. Accordingly, the E1/2 values, which are the average of E1red and E1ox, are used for the discussion of the electrochemical properties of the substrates. The E1/2 values for 2a–c and 3c,d are cathodically shifted compared to that of fullerene, indicating that the electron densities of the fullerene moieties of 2 and 3c,d are higher than that of C60 itself. The increased electron densities are the result of saturation of a carbon–carbon double bond of the fullerenes via the attachment of the arylethynyl groups.11a,14,15 Thus, it is clear that the fullerene moieties of 2 and 3c,d are much more electron-rich than C60 itself in the ground state. The fullerene moieties of 2 attract electrons from the arylethynyl groups, even though electron-withdrawing carboranes are attached to the para position of the phenyl groups, and this experimental result is in good agreement with the fact that fullerene is an electron-attracting carbon cluster. It is interesting to consider the order of redox potentials of the fullerene moieties of 2a–c and 3c,d
(Table 4). The order of the cathodic shift of the E1/2 values is as follows: X =
ortho-carborane ≥
meta-carborane > para-carborane > H > Me. This order indicates that the fullerene moieties of 3c,d
(X = H and Me) are more difficult to reduce, i.e. more electron-rich, than those of the carborane derivatives 2, since Me is an electron-donating group and carboranes are electron-withdrawing groups. As mentioned above, the electron densities of the fullerene moieties of 2 and 3c,d are higher than that of C60 itself, but among them, the E1/2 values of 2a–c exhibit lower cathodic shifts in comparison with those of 3c,d, as a result of the electron-withdrawing character of the carboranes.
Table 4 Redox potentialsa of C60, 2 and 3c,d
| Compound |
X |
E
1
red
|
E
1
ox
|
E
1/2
b
|
|
Potentials in mV vs. ferrocene/ferrocenium measured in o-dichlorobenzene.
Of the three quasi-reversible redox peaks, only the E1/2 [1/2(E1red
+
E1ox)] values for the first peaks are listed, the other peaks are shown in the ESI.
|
| C60 |
— |
−1103 |
−631 |
−867 |
|
2a
|
o-Carborane |
−1242 |
−552 |
−897 |
|
2b
|
m-Carborane |
−1210 |
−581 |
−896 |
|
2c
|
p-Carborane |
−1193 |
−615 |
−904 |
|
3c
|
H |
−1086 |
−752 |
−919 |
|
3d
|
Me |
−1108 |
−766 |
−937 |
Role of the fullerene moiety in SHG activity
SHG activities are generally influenced by the dipole moments in the ground and excited states. The dipole moments are affected by intramolecular charge transfer. In general, it is expected that charge transfers make β values large. A good correlation between Hammett σp values and β values likely indicates that, in the excited state, the fullerene moieties of 2 and 3c,d act as donors, although they exhibit electron-accepting properties in the ground state, as often mentioned in the literature. Therefore, 2a,c and 3a,b exhibit higher SHG activities than 3c,f. There has been no previous report that fullerene acts as an electron-donating group, to the best of our knowledge.
There is a sp3-carbon between the substituted phenylacetylene moieties and fullerene π-systems in 2 and 3. Usually there are no strong interactions between the two substituents via the sp3-carbon. As mentioned above, since a clear substituent effect was observed for 2a–c and 3c,d, there must be an interaction between the π-spacer and the fullerene cages through the sp3-carbon atom. Wudl and co-workers have already observed such a phenomenon, known as periconjugation, wherein their is an expansion of π-conjugation between π-systems on fullerenes and another π-systems.16,17 Periconjugation is presumably operative in the present system, as shown in Fig. 3. Additionally, longer conjugation systems usually give higher β values. In the case of 2 and 3, expansion of the π-conjugation systems can account for both the E1/2 and β values. To the best of our knowledge, 2 and 3 are the first examples, in which enhancement of the NLO activity was observed through an interaction between C60 and a π-spacer.
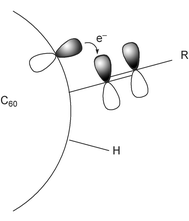 |
| | Fig. 3 Interaction between the fullerene π-system and the arylethyne moiety through the sp3 carbon in 2 and 3. | |
Higher β values due to carborane moieties
The final question is why 2, containing carborane, exhibit higher β values in comparison with 3a,b, bearing electron-withdrawing groups, and why the para-carborane derivative 2c exhibits the highest β value among the carborane-appended molecules. It is well known that closo-carboranes are electron-deficient boron clusters with highly polarizable σ-aromatic character, which means delocalization of electrons in the cages. Accordingly, not only in NLO materials,7 but also in ordinary organic reactions,18,19closo-carboranes are often used as electron acceptors. For example, the pKa values of C–H bonds in simple closo-carboranes are shown in Scheme 4. The order of C–H acidity in simple closo-carboranes is ortho > meta
≫
para,20i.e. the ortho-carborane has the most acidic proton, or, in other words, the greatest electron-attracting ability among these isomers. However, the order of β values is ortho < meta
≪
para-substituted derivative, the para-carborane derivative exhibiting the highest β value among 2a–c
(Scheme 4).
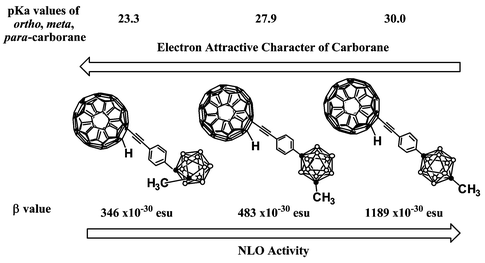 |
| | Scheme 4 | |
We think that the σ-aromaticity of the carborane structural framework is key to understanding the phenomena mentioned above. The σ-aromaticity makes carboranes very stable by delocalizing electrons on the cage, even though they are electron deficient. This character also allows the two carbons in carborane cage to interact with each other very easily and occurs even in the case of para-carborane, in spite of the distance between the carbon atoms.21,22 In the case of 2, such an interaction affects the β values because one of the two carbons combines with the phenylacetylene–fullerene moiety. We assume that, during the excitation process, the electron density at the carbon on carborane cage which is linked to fullerene through the π-spacer increases due to the electron-donating ability of C60, leading to significant differences in electron density between the carbon near the donor and the other carbon. The electronegativities of carbon (2.5) and boron (2.0) indicate that the electron-attracting ability of carbon in carborane is higher than that of boron. Consequently, charge transfer occurs from the electron-rich carbon to the other carbon in the exited state. Such an interaction between two carbons by σ-aromaticity produces a new conjugation system in carborane in addition to the phenylacetylene–C60
π-conjugation system. Thus, compounds 2 possess not only a π-conjugation system due to phenylacetylene and C60, but also a σ-conjugation system due to the carboranes. It has been shown that there are interactions between the carbon in carborane and aromatic substituents on it.21 Taking these results together, it is now clear that the conjugation system of 2 is longer than that of 3. It is widely accepted that longer π-conjugation systems make β values higher. Consequently, the difference between the carboranes in 2 and the more common acceptors in 3 is the presence of σ-aromaticity; compounds 2, having longer π- and σ-conjugations, exhibit higher SHG activities than compounds 3.
Finally, it should be mentioned that the NLO activity of the para-carborane derivative 2c is higher than those of 2a,b. The two carbons in ortho-carborane are adjacent, whereas the two carbons of para-carborane occupy the 1- and 12-positions in the icosahedral cage. The difference between the three carborane isomers is the distance between the two carbons in their cages. As mentioned above, these carbons interact with each other through σ-aromaticity and a more extended conjugation system leads to higher NLO activity. Among compounds 2, the para-carborane derivative has the longest distance between these two carbons. The longer the distance between carbons, the longer the σ-conjugation system and, hence, the higher the β value. This is why the order of the β values for compounds 2 is para
≫
meta > ortho-carborane derivatives. It is now clear that carboranes are special acceptors which not only have electron-attracting ability, but also act as prolonged conjugation systems.
Conclusion
The fullerene–acceptor systems 3a,b give higher β values (SHG activities 350 and 361 × 10−30 esu, respectively) than the fullerene–donor systems 3c–f
(137, 127, 72 and 20 × 10−30 esu, respectively). Furthermore, 2a–c, having carboranes at the para position of the phenyl ring of the fullerene–arylethyne hybrids exhibit significantly higher β values (346, 483 and 1189 × 10−30 esu, respectively) than those for 3a–f. Among the ortho-, meta- and para-carborane derivatives 2a–c, the para derivative 2c shows an exceptionally high β value of 1189 × 10−30 esu. A good correlation was obtained between the β values of compounds 3 and the Hammett σp values of their para substituents, X, indicating that the fullerene moieties of 3 act as electron-donating groups in the excited state. The redox properties of 2 and 3c,d indicate that the fullerene moiety acts as an acceptor in the ground state, which is in good agreement with the accepted view that fullerene is an electron-attracting carbon cluster. The high SHG activities of the carborane–fullerene dyads 2 are most probably due to the electron-donating character of fullerene in the excited process, together with the electron-withdrawing nature of the carborane clusters. The exceptionally high β value for the para-carborane derivative 2c is due to the synergistic effect of the prolonged conjugation and electron-withdrawing effect of para-carborane.
References
-
(a)
P. N. Prasad and D. J. Williams, Nonlinear Optical Effects in Molecules and Polymers, Wiley, New York, 1991 Search PubMed;
(b)
P. N. Prasad, Nonlinear Optical Properties of Organic Materials, Plenum, New York, 1991 Search PubMed.
-
(a)
P. Harper and B. Wherrett, Nonlinear Optics, Academic Press, New York, 1997 Search PubMed;
(b)
B. E. A. Saleh and M. C. Teich, Fundamentals of Photonics, Wiley, New York, 1991 Search PubMed.
- See, for example:
(a) S. R. Marder, C. B. Gorman, B. G. Tiemann and L.-T. Cheng, J. Am. Chem. Soc., 1993, 115, 3006 CrossRef CAS;
(b) S. R. Marder, D. N. Beratan and L.-T. Cheng, Science, 1991, 252, 103 CAS.
- M. Lamrani, R. Hamasaki, M. Mitsuishi, T. Miyashita and Y. Yamamoto, Chem. Commun., 2000, 1595 RSC.
-
G. A Olah, K. Wade and R. E. Williams, Electron Deficient Boron and Carbon Clusters, Wiley, New York, 1990 Search PubMed.
-
(a) D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195 CrossRef CAS;
(b) Q. Xie, E. Pérez-Cordero and L. Echegoyen, J. Am. Chem. Soc., 1992, 114, 3978 CrossRef;
(c) F. Zhou, C. Jehoulet and A. J. Bard, J. Am. Chem. Soc., 1992, 114, 11004 CrossRef CAS;
(d) Y. Ohsawa and T. Saji, J. Chem. Soc., Chem. Commun., 1992, 781 RSC;
(e) T. D. Ros, M. Prato, M. Carono, P. Ceroni, F. Paolucci and S. Roffia, J. Am. Chem. Soc., 1998, 120, 11645 CrossRef CAS;
(f) N. Martin, L. Sanchez, B. Illescas and I. Perez, Chem. Rev., 1998, 98, 2527 CrossRef CAS.
- D. M. Murphy, D. M. P. Mingos and J. M. Forward, J. Mater. Chem., 1993, 3, 67 RSC.
- Y Wang and L.-T. Cheng, J. Phys. Chem., 1992, 96, 1530 CrossRef CAS.
- D. Zhu, C. Zhu, Y. Xu, C. Long, Y. Liu, M. Han, Y. Yao, X. Zhao and X. Xia, Thin Solid Films, 1996, 284–285, 102 CrossRef CAS.
- S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Synthesis, 1980, 627 CrossRef CAS.
-
(a) K. Komatsu, Y. Murata, N. Takimoto, S. Mori, N. Sugita and T. S. M. Wan, J. Org. Chem., 1994, 59, 6101 CrossRef CAS;
(b) Y. Murata, K. Motoyama, K. Komatsu and T. S. M. Wan, Tetrahedron., 1996, 52, 5077 CrossRef CAS.
-
R. Taylor, Lecture Notes on Fullerene Chemistry, Imperial College Press, London, 1999 Search PubMed.
- J. Catalán and J. Elguero, J. Am. Chem. Soc., 1993, 115, 9249 CrossRef CAS.
- M. Maggini, A. Karlsson, G. Scorrano, G. Sandonà, G. Farnia and M. Prato, J. Chem. Soc., Chem. Commun., 1994, 589 RSC.
- M. A. Herranz, B. Illescas, N. Martín, C. Luo and D. M. Guldi, J. Org. Chem., 2000, 65, 5728 CrossRef CAS.
- F. Wudl, T. Suzuki and M. Prato, Synth. Met., 1993, 59, 297 CrossRef CAS.
-
(a) M. Eiermann, R. C. Haddon, B. Knight, Q. Chan Li, M. Maggini, N. Martin, T. Ohno, M. Prato, T. Suzuki and F. Wudl, Angew. Chem., Int. Ed. Engl., 1995, 34, 1591 CrossRef CAS;
(b) T. Ohno, N. Martin, B. Knight, F. Wudl, T. Suzuki and H. Yu, J. Org. Chem., 1996, 61, 1306 CrossRef CAS;
(c) B. Knight, N. Martin, T. Ohno, E. Orti, C. Rovira, J. Veciana, J. Vidal-Gancedo, P. Viruela, R. Viruela and F. Wudl, J. Am. Chem. Soc., 1997, 119, 9871 CrossRef CAS.
-
(a) H. Nakamura, K. Aoyagi and Y. Yamamoto, J. Org. Chem., 1997, 62, 780 CrossRef CAS;
(b) H. Nakamura, K. Aoyagi, B. Singaram, J. Cai, H. Nemoto and Y. Yamamoto, Angew. Chem., Int. Ed. Engl., 1997, 36, 367 CrossRef CAS;
(c) H. Nakamura, K. Aoyagi and Y. Yamamoto, J. Am. Chem. Soc., 1998, 120, 1167 CrossRef CAS.
-
(a) Y. Endo and Y. Taoda, Tetrahedron Lett., 1999, 40, 9073 CrossRef CAS;
(b) Y. Endo, T. Sawabe and Y. Taoda, J. Am. Chem. Soc., 2000, 122, 180 CrossRef CAS.
- A. I. Shatenshtein, L. I. Zakharkin, E. S. Petrov, E. A. Yakovleva, F. S. Yakushin, Z. Vukmirovich, G. G. Isaeva and V. N. Kalinin, J. Organomet. Chem., 1970, 23, 313 CrossRef CAS.
- M. A. Fox, J. A. H. MacBride, R. J. Peace and K. Wade, J. Chem. Soc., Dalton Trans., 1998, 401 RSC.
-
(a) D. G. Allis and J. T. Spencer, J. Organomet. Chem., 2000, 614, 309 CrossRef;
(b) D. G. Allis and J. T. Spencer, Inorg. Chem., 2001, 40, 3373 CrossRef CAS;
(c) J. Taylor, J. Caruso, A. Newlon, U. Englich, K. Ruhland-Senge and J. T. Spencer, Inorg. Chem., 2001, 40, 3381 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2003 |
Click here to see how this site uses Cookies. View our privacy policy here. 
![Cyclic voltammogram of 2a
(5 ×10−3 M) and C60
(2.5 × 10−3 M) in o-dichlorobenzene {supporting electrolyte 0.1 M n-Bu4NClO4; working electrode glassy carbon; counter electrode Pt wire; reference electrode Ag/0.01 N AgNO3 in acetonitrile with 0.1 M n-Bu4NClO4 [E1/2(ferrocene/ferrocenium)
= 180 mV]; scan rate 100 mV s−1}.](/image/article/2003/JM/b205258j/b205258j-f2.gif)