ICP-MS determination of Pt, Pd and Rh in airborne and road dust after tellurium coprecipitation
M. B.
Gómez
,
M. M.
Gómez
* and
M. A.
Palacios
Departamento de Química Analítica, Facultad de CC. Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: mmgomez@quim.ucm.es; Fax: 34913944329
First published on 20th November 2002
Abstract
A method has been developed for the simultaneous determination of Pd, Pt and Rh (PGE) in environmental airborne and road dust samples by tellurium coprecipitation and ICP-MS. The Te coprecipitation was applied after digestion of the sample with aqua regia–HF in a microwave oven. This separation method removes more than 95% of the elements producing mass interference in PGE determination by ICP-MS. The methodology was validated with reference road dust samples CW7 and CW8. The detection limits are 0.3, 0.6 and 0.8 pg m−3 for Pt, Pd and Rh in airborne particulate matter, and 1, 1 and 0.4 ng g−1 for Pt, Pd and Rh in road dust. Application of the isotopic dilution method for Pt and Pd after their coprecipitation improves the results obtained for road dust samples. Rh (monoisotopic element) analysis was carried out by external calibration after Te coprecipitation.
Introduction
Research on Pt in environmental samples such as road dust, water, plants, micro-organisms, etc., has proceeded in parallel with the Pt contamination produced by the implementation of Pt automotive catalysts in the mid-1980s and the development of very sensitive methods for Pt determination using inductively coupled plasma mass spectrometry (ICP-MS) and cathodic stripping voltammetry (CSV) techniques.1–8Pt is increasingly being replaced by Pd in catalyst technology, and Rh is now used in the catalyst for NOx reduction. Cytotoxicity, mutagenic effects, bioaccumulation capability and other undesirable effects in living organisms have already been reported for environmental Pd. Therefore, analogous research to that performed for Pt should be carried out.9–12 The effect of Rh in the environment is practically unknown. There is a need to develop precise and sensitive methods for the determination of Pd and Rh, especially in those samples where interaction with living organisms is evident, such as airborne and road dust. However, it is difficult to establish a robust methodology for Pd and Rh in a similar way to that used for Pt.
ICP-MS is the only technique for routine Pt, Pd and Rh determination at the ultra-trace level. However, mass interferences produced by matrix components may hamper the direct determination of these elements.13,14 To obtain reliable data for Pd and Rh, matrix separation is necessary. A successful preconcentration/matrix separation method for determination of PGE in geological and some environmental samples is the NiS–fire assay method.15–17 However, the high blank level and the incomplete recovery of PGE hinder its application to samples with very low PGE content such as airborne and road dust.18–22 The tellurium coprecipitation of these elements after fire assay or alkaline fusion has been applied for improving the preconcentration/matrix separation performance.
This paper describes a Te coprecipitation method, applied after acid digestion of the sample, for removal of mass interferences in Pd, Rh and Pt determination by ICP-MS in airborne and road dust. For Pt and Pd analysis, the ability to carry out the analysis by isotopic dilution-ICP-MS (ID-ICP-MS) after Te coprecipitation has been evaluated in order to improve the accuracy of the results.
Experimental
Reagents and apparatus
Te (1000 mg L−1) stock solution was prepared from TeIV chloride (Acros Organics) in 2 mol L−1 HCl and stored in a PTFE bottle. SnCl2 (1 mol L−1) was prepared from SnII chloride (Merck, Darmstadt, Germany) in 6 mol L−1 HCl. Pt, Pd, Rh, internal standards and interferent elements standard solutions of 1000 mg L−1 (Merck, Darmstadt, Germany) were employed. Concentrated HNO3 and HCl (Panreac, Barcelona, Spain) were purified by sub-distillation in a PTFE distiller BSB-939IR (Berghof, Eningen, Germany).HF was Suprapur grade (Merck, Darmstadt, Germany). High-purity deionised water was obtained from a Milli-Q system (Millipore, Molsheim, France).
Enriched 108Pd and 194Pt standard metals were obtained from Cambridge Isotope Laboratories (Woburn, MA, USA) and dissolved in aqua regia (HCl∶HNO3, 3∶1) to prepare the stock solutions (standardised by reverse isotopic dilution) of 51.36 µg g−1 (uncertainty23 for k = 2 of 1.20) and 60.25 µg g−1 (uncertainty for k = 2 of 1.25), respectively, for three replicates. Table 1 shows the isotopic composition of natural and enriched standards for the Pt and Pd isotopes employed.
| Abundance (%) | Isotopes | ||||||
|---|---|---|---|---|---|---|---|
| 105Pd | 106Pd | 108Pd | 194Pt | 195Pt | 196Pt | ||
| Natural element | 22.33 | 27.33 | 26.46 | 32.90 | 33.80 | 25.30 | |
| Enriched element | 0.63 | 2.84 | 94.19 | 91.46 | 6.75 | 1.60 | |
A Q-ICP-MS (HP-4500, Agilent Technologies, Yokogawa Analytical System, Tokyo, Japan), equipped with a Babington-type nebulizer, a Fassel torch and a double-pass Scott-type spray chamber cooled by a Peltier system, was employed for the measurements.
A microwave oven system MSP-1000 (CEM, Matthews, NC, USA) with a maximum power output of 1000 W was employed for sample mineralization.
Samples and sampling collection
The sampling of airborne matter was performed with a PM-10 collector (Partisol 2000, 1 m3 h−1) with mixed cellulose ester filters (0.45 µm, 47 mm, Millipore) over 48 h sampling time. For road dust sampling, a vacuum cleaner (Nilfisk, GM-80) was employed over a surface of about 10 m2.Airborne and road dust was collected at selected places along the M-30 urban highway and in downtown Madrid. CW 7 and CW 8 road dusts were collected from the walls of the ventilation system of the Tanzenberg tunnel, located 50 km north of the city of Graz (Austria) in 1994 (CW7) and 1998 (CW8), respectively.24
Sample preparation
Airborne dust. Filters with the collected airborne dust were dissolved with 8 mL of aqua regia in the microwave oven at 60% power during 15 min. After cooling, 1 mL of HF was added and mineralised again for 15 min at 60% power. The digested samples were transferred to PTFE vessels and heated to dryness on a hot plate. 2 mL of HCl were added and heated to dryness (this step was repeated twice). Finally, the digested samples were diluted to 25 mL with 1 mol L−1 HCl.Road dust. Only the <63 µm fraction was analysed. 0.1–0.2 g were digested in a similar way to the airborne dust samples, but it was necessary to repeat the second microwave oven step and to add 3 mL of HF before heating the sample to dryness, in order to ensure total digestion of the sample.
CW 7 and CW 8 dust. 0.2 g of sample was heated in a porcelain crucible for 2 h at 450 °C to eliminate the organic matter. After cooling, the sample was transferred to a microwave vessel and mineralised following the road dust preparation procedure.
Te coprecipitation procedure. 2 mL of Te solution (1000 mg L−1) was added to the 25 mL solution of mineralised samples and heated to boiling. 2 mL of SnII chloride was slowly added and boiled for approximately 10 min until the black precipitate was thoroughly coagulated. 1 mL more of Te solution was added and the mixture boiled again for further coagulation of the precipitate. After cooling, the precipitate was filtered under vacuum through 0.45 µm cellulose ester filters and rinsed with hot 1 mol L−1 HCl. The filter and the precipitate were transferred to PTFE vessels, and the black precipitate was dissolved with the minimum volume of concentrated HNO3 until total dissolution. The resulting solution was evaporated to dryness on a hot plate and the residue was dissolved in 0.25 mL aqua regia and heated for a few seconds. The final volume was made up to 25 mL with Milli-Q water.
Analysis by ICP-MS and external calibration
The operating conditions for ICP-MS analysis by external calibration are summarised in Table 2. The selected internal standards were 10 µg L−1 of Ru for 103Rh and 105Pd and 10 µg L−1 of Ir for 195Pt. No important memory effects for PGE were observed using 2% (v/v) HNO3 as rinse solution and a wash time of 90s.| Plasma— | |
| RF power | Forward: 1200 W |
| Reflected: <5 W | |
| Ar flow-rate | Coolant: 15 L min−1 |
| Nebulizer: 0.95–1.2 L min−1 | |
| Auxiliary: 1.3 L min−1 | |
| Solution uptake rate | 1 mL min−1 |
| Temperature spray chamber | 5 °C |
| Acquisition parameters— | |
| Measurement mode | Peak jumping (peak height) |
| 103Rh, 105Pd, 106Pd, 108Pd, 194Pd, 195Pt, 194Pt, 63Cu, 65Cu, 85Rb, 88Sr, 89Y, 90Zr, 95Mo, 179Hf, 206Pb, 64Zn, 110Cd | |
| Integration time | 0.1 s per point (external calibration) |
| 106Pd, 108Pd, 0.5 s per point; 194Pt, 195Pt, 0.6 s per point (isotopic dilution) | |
| Points per peak | 3 |
| Wash time | 90 s |
| Number of replicates | 5 |
| Internal standards | 101Ru, 191I |
Analysis by ICP-MS and isotopic dilution (ID)
A known amount of the isotope-enriched stock solution of Pt and Pd was added to the samples before mineralization.25Table 2 shows the instrumental conditions for ICP-MS analysis by ID.
The concentration of the analytes was calculated from the following equation:26
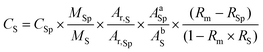
To minimise the uncertainty introduced by the propagation error through the isotope dilution equation, the random error propagation was applied in order to select the optimum isotopes and the optimum spike to sample ratio. The selected isotope ratios were 195/194 for Pt and 106/108 for Pd, which present the higher optimum values for Rm: 0.27 and 0.18, respectively. The amount of spikes added to the analysed samples are in the ranges of 20–80 ng of 194Pt and of 2–30 ng of 108Pd.
R m was corrected for instrumental mass bias with a solution of 10 µg L−1 of natural Pd and Pt.
Detector dead time did not need to be taken into account as it is automatically corrected by the software of the instrument.
Results and discussion
Te coprecipitation and interferents separation in PGE analysis by ICP-MS
A previous interference study carried out for airborne and road dust samples showed that Hf isotopes interfere in Pt isotope determination by HfO+ formation. However, the interference is in the range 10–20% of the analyte signal and mathematical corrections can be applied. Rh, a monoisotopic element, suffers from interference by ArCu+, Pb2+, SrO+ and RbO+ in a 30–75% range. Pd is subject to interference by ArCu+, ArZn+, YO+, Cd+, MoO+ and ZrO+ at such a high level that determination is not possible.13,14The reduction of TeIV to Te0 by stannous chloride induces PGE precipitation.20,27 The ability of Te coprecipitation to separate Pd, Pt and Rh from the elements that cause mass interferences has been studied.
Table 3 shows the average content of interferent elements in some dust samples and their recovery in the Te precipitate. For most of the interferents and in most of the samples, the recovery in the Te precipitate is lower than 2%. Although most Cu and Y were removed (>95%), the remaining fraction in the precipitate produces a signal contribution to 105Pd of about 1–10% from YO+ (oxide formation rate, RYO+/Y+ = 0.025) and 1–15% from ArCu+. This made it advisable to correct mathematically the contribution of these isobaric mass interferences to the signal of 105Pd isotope14 used in the determination by external calibration.
| CW 7 | Road dust | Airborne dust | ||||
|---|---|---|---|---|---|---|
| a n.d.: not detected. | ||||||
| Interferent element (mass interference) | Content/µg g−1 | Recovery (%) | Content/µg g−1 | Recovery (%) | Content/ng m−3 | Recovery (%) |
| Cu (ArCu+) | 160 | 2 | 473 | 5 | 150 | 4.3 |
| Rb (RbO+) | 65 | n.d. | 184 | n.d. | 2.5 | n.d. |
| Sr (SrO+) | 175 | 0.08 | 290 | 0.01 | 15.7 | 1.3 |
| Y (YO+) | 7.5 | 0.6 | 4.9 | n.d. | 0.7 | 1.4 |
| Hf (HfO+) | 0.6 | n.d | 3.7 | n.d. | 0.13 | n.d. |
| Pb (Pb2+) | 1041 | 0.02 | 1640 | 0.01 | 304 | 0.3 |
| Zn (ArZn+) | 134 | 0.1 | 797 | 0.1 | 202 | 0.15 |
| Zr (ZrO+) | 27.9 | n.d. | 89 | 0.01 | 4.8 | 1.0 |
| Mo (MoO+) | 38 | n.d. | 9.5 | 0.05 | 3.6 | 0.5 |
| Cd (Cd+) | 2.6 | 4 | 1.1 | n.d | 2.0 | n.d |
Mathematical corrections were not necessary for Rh.
Recoveries in the Te precipitate for the analysed PGE were carried out in triplicate by spiking the samples (before digestion) with 2.5 µg of Pt, Pd and Rh, and were in the range 70–80% for Pd, 80–90% for Rh and 40–60% for Pt. The low recovery obtained for Pt and the low level of Hf interference made advisable the direct determination of Pt (without Te coprecipitation) by mathematical correction of HfO+ interference (RHfO+/Hf+ = 0.03).
Analysis of PGEs in dust samples
Table 4 shows the results obtained for PGE in the selected samples by external calibration and isotope dilution analysis.| Material | Element | Content Te—EC | Content Te—ID | Content IA |
|---|---|---|---|---|
| a Results given as ± SD (n = 3). b Sampling points: A.I. = Avenida de la Ilustración (M-30 urban highway); E. A. = Escuelas Aguirre (downtown); M.M. = Marqués de Monistrol (M-30 urban highway). c Calculated by direct analysis (without Te coprecipitation), applying mathematical corrections. IA: Interlaboratory analysis. d Certified value. | ||||
| CW7/ng g−1 | Pt | 50.6 ± 1.3c | 55 ± 2 | 55 ± 8 |
| Pd | 6.7 ± 2.1 | 4.3 ± 0.2 | 4.0 ± 1.3 | |
| Rh | 10.0 ± 1.5 | — | 10.3 ± 1.4 | |
| CW8/ng g−1 | Pt | 81.4 ± 6.7c | 83 ± 4 | 81.3 ± 6.2d |
| Pd | 5.5 ± 1.2 | 5.1 ± 0.7 | 5.5 ± 1.8d | |
| Rh | 13.0 ± 1.5 | — | 12.8 ± 2.0d | |
| Airborne A.I./pg m−3 | Pt | 15 ± 2c | — | — |
| Pd | 5.1 ± 0.9 | — | — | |
| Rh | 27 ± 1 | — | — | |
| Airborne E. A./pg m−3 | Pt | 19 ± 1c | — | — |
| Pd | 32 ± 1 | — | — | |
| Rh | 9.1 ± 0.2 | — | — | |
| Road dust A. I./ng g−1 | Pt | 339 ± 3c | 377 ± 5 | — |
| Pd | — | 75 ± 2 | — | |
| Rh | 64 ± 1 | — | — | |
| Road dust M. M./ng g−1 | Pt | 144 ± 19c | 198 ± 7 | — |
| Pd | — | 26 ± 3 | — | |
| Rh | 44 ± 1 | — | — | |
105Pd isotope was selected for external calibration due to the lack of any contribution of Cd+ interference in these samples. 106Pd isotope was selected for ID in order to avoid the molecular interferences ArCu+ and YO+. However, in some samples it was necessary to correct the isobaric mass interference of Cd+ in the 106Pd and 108Pd isotope signals.
Rh cannot be determined by ID methodology because it is monoisotopic and there are not any convenient radioactive isotopes available.
Pt was analysed directly without Te coprecipitation in external calibration analysis. The differences in the results for Pt in road dust, 10% for A.I. and 27% for M.M., between external calibration without Te coprecipitation and ID with Te coprecipitation, are unknown. A possible explanation could be an overestimation of error in the mathematical correction of HfO+ interference in external calibration.
The PGE concentration obtained in the participation in the feasibility and certification campaigns of CW7 and CW8, respectively, and for both types of analysis (EC and ID) agree with the certified value for CW8 and with the average of the selected interlaboratory results for CW7.24 In the case of ID, this agreement in the results indicates a good equilibration between spike and sample isotopes. The standard deviations obtained in the ID method are lower than the external calibration, indicating more precise methodology.
Blank levels and detection limits
Blank samples were prepared following the same procedure used for the real samples. The calculation of concentrations was based on the analysis of one filter of 48 m3 of air for airborne matter and 0.2 g of sample for road dust. The blank levels obtained were about 2.2 pg m−3 for airborne matter and 3.4 ng g−1 for road dust. The detection limits obtained were of 0.3, 0.6 and 0.8 pg m−3 for Pt, Pd and Rh, respectively, in airborne matter and 1, 1 and 0.4 ng g−1 for Pt, Pd and Rh, respectively, in road dust.Conclusions
Pd can be determined in dust samples at ultra-trace level after Te coprecipitation by both external calibration with the 105Pd isotope and ID with the 106Pd isotope. Although removal of the main interferent elements after precipitation is higher than 95%, the contributions of ArCu+ and YO+ in 105Pd and of Cd+ in 106Pd have to be checked in the samples, and mathematically corrected if necessary.Rh is not subject to interference after Te coprecipitation and determination by external calibration gives acceptable results.
Pt is the element which suffers the least interference after Te coprecipitation. However, the low recovery obtained makes advisable its direct determination after acid digestion of the sample and mathematical correction of the HfO+ contribution.
However, the Te-coprecipitation-ID-ICP-MS methodology is not completely free of drawbacks when it is applied to airborne samples, due to the very low PGE level and the need to add a weighed amount of spike isotope, which means that more research is necessary.
Acknowledgements
This work was financially supported by the European Union under the Environmental and Climate Program (CEPLACA Project, ENV4-CT97-0518).References
- M. E. Farago, P. Kanavagh, R. Blanks, J. Kelly, G. Kazanstzis, I. Thornton, P. R. Simpson, J. M. Cook, S. Parry and G. E. M. Hall, Fresenius’ J. Anal. Chem., 1996, 354, 660 CAS.
- B. Gómez, M. A. Palacios, M. Gómez, J. L. Sánchez, G. M. Morrison, S. Rauch, C. McLeod, R. Ma, S. Caroli, A. Alimonti, F. Petrucci, B. Bocca, P. Schramel, M. Zischka, C. Petterson and U. Wass, Sci. Tot. Environ., 2002, 299(3), 1 Search PubMed.
- S. Lustig, S. Zang, B. Michalke and P. Schramel, Fresenius’ J. Anal. Chem., 1997, 357, 1157 CrossRef CAS.
- M. Moldovan, S. Rauch, M. M. Gómez, M. A. Palacios and G. M. Morrison, Wat. Res., 2001, 35(17), 4175 Search PubMed.
- O. Nygren, G. T. Vaughan, T. M. Florence, G. M. P. Morrison, L. M. Warner and L. S. Dale, Anal. Chem., 1990, 62, 1637 CrossRef CAS.
- J. Begerow, M. Turfeld and L. Dunnemann, J. Anal. At. Spectrom., 1997, 12, 1095 RSC.
- M. Krachler, A. Alimonti, F. Petrucci, K. J. Irgolic, F. Forastiere and S. Caroli, Anal. Chim. Acta, 1998, 363, 1 CrossRef CAS.
- S. Rauch and G. M. Morrison, Sci. Tot. Environ., 1999, 235(1-3), 261 Search PubMed.
- M. A. Palacios, M. M. Gómez, M. Moldovan, G. Morrison, S. Rauch, C. McLeod, R. Ma, J. Laserna, P. Lucena, S. Caroli, A. Alimonti, P. Schramel, S. Lustig, U. Wass, B. Stenbom, M. Luna, J. C. Saenz, J. Santamaría and J. M. Torrens, Sci. Tot. Environ., 2000, 257, 1 Search PubMed.
- CEPLACA. Assessment of Environmental Contamination Risk by Platinum, Rhodium and Palladium from Automobile Catalyst (ENV4-CT97-0518). Final Report, 2001.
- J. Bünger, J. Stork and K. Stahlder, Int. Arch. Occup. Environ. Health, 1996, 69, 33 CrossRef CAS.
- J. C. Wataha and C. T. Hanks, J. Oral Rehabilitation, 1996, 23(5), 309 Search PubMed.
- M. B. Gómez, M. M. Gómez, J. L. Sánchez, R. Fernández and M. A. Palacios, Sci. Tot. Environ., 2001, 269(1-3), 131 Search PubMed.
- M. B. Gómez, M. M. Gómez and M. A. Palacios, Anal. Chim. Acta, 2000, 404, 285 CrossRef CAS.
- R. Juvonen, E. Kallio and T. Lakomaa, Analyst, 1994, 119, 617 RSC.
- M. Zischka and W. Wegscheider, in Anthropogenic Platinum Group Element Emissions—Their Impact on Man and Environment eds. F. Zereini and F. Alt, Springer Verlag, Berlin and Heidelberg, 2000, pp 201 Search PubMed.
- G. Ravizza and D. Pyle, Chem. Geol., 1997, 141(3-4), 251 CrossRef CAS.
- J. Enzweiler and P. J. Potts, Analyst, 1995, 120, 1391 RSC.
- S. Yali, G. Xiyun and D. Andao, Spectrochim. Acta, Part B, 1998, 53, 1463 CrossRef.
- J. Amossè, Geostand. Newsl., 1998, 22(1), 93 Search PubMed.
- K. Oguri, G. Shimoda and Y. Tatsumi, Chem. Geol., 1999, 157(3-4), 189 CrossRef CAS.
- X. Jin and H. Zhu, J. Anal. At. Spectrom., 2000, 15, 747 RSC.
- Quantifying Uncertainty in Analytical Measurements, EURACHEM, London, UK, 1995 Search PubMed.
- P. Schramel, M. Zischka, H. Muntau, B. Stojanik, R. Dams, M. Gómez and Ph. Quevauviller, J. Environ. Monit., 2000, 2, 443 RSC.
- K. G. Heumann in: Inorganic Mass Spectrometry: Isotope Dilution Mass Spectrometry, eds. F. Adams, R. Gijbels and R. Van Grieken, Wiley, New York, 1988, pp. 301 Search PubMed.
- J. L. García Alonso, Anal. Chim. Acta, 1995, 312, 57 CrossRef.
- A. S. Meyer Jr. and G. H. Ayres, J. Am. Chem. Soc., 1955, 77, 2671 CrossRef.
| This journal is © The Royal Society of Chemistry 2003 |