Separation of picogram quantities of Cr(III) and Cr(VI) species in aqueous solutions and determination by graphite furnace atomic absorption spectrometry
Sanna
Latva
*,
Jonna
Jokiniemi
,
Sirpa
Peräniemi
and
Markku
Ahlgrén
University of Joensuu, Department of Chemistry, P.O.Box 111, FIN-80101 Joensuu, Finland
First published on 2nd December 2002
Abstract
A procedure was developed for the separation and independent determination of picogram quantities of Cr(III) and Cr(VI) in aqueous solution. Cr(VI) was adsorbed onto iron-loaded activated charcoal (FeC*), while Cr(III) was left in the filtrate and was measured directly by graphite furnace atomic absorption spectrometry (GFAAS). The Cr(VI) adsorbed on FeC* was released with 2.0 M HCl before measurement by GFAAS. The detection limits were 7.0 pg for total Cr, 1.9 pg for Cr(III) and 6.8 pg for Cr(VI). The recoveries were 98 ± 4 for total Cr, for Cr(III) 97 ± 3 and for Cr(VI) 101 ± 7. The procedure was tested with the Ground Water and Waste Pollution Control Check Standard WP-15 spiked with Cr(VI) and with samples from the plating industry.
1. Introduction
Chromium enters natural waters in effluents from electroplating and tanning processes, production of pigments and wood preservatives, sanitary landfill leaching and water-cooling towers.1,2 Chromium exists primarily in two oxidation states: trivalent Cr(III) and hexavalent Cr(VI). Cr(III) probably exists in natural waters in hydrolysed Cr(H2O)4(OH)2+ form, in complexes and even adsorbed on colloidal matter. The Cr(VI) species most frequently found in natural waters are chromate (CrO42−) and dichromate (Cr2O72−) ion, the particular form depending on the pH of the solution.2,3 Chromium(III) is an essential trace element for humans, required for the maintenance of normal glucose, cholesterol and fatty acid metabolism. Cr(VI), in turn, is highly toxic; its acute toxic effects include immediate cardiovascular shock, with later effects on kidney, liver and blood-forming organs.4,5 Owing to the different toxicities of Cr(VI) and Cr(III), it is important to determine them separately in addition to the total chromium content.6 Information about the oxidation state of chromium is also very important for many industrial processes and waste water purification methods.Ion exchange,7,8 coprecipitation9,10 and complex formation with subsequent solvent extraction9,11 have all been used for the separation of Cr(VI) and Cr(III) species. A major drawback of many of the procedures is that only one chromium species is measured, and the other is determined as the difference between total chromium, determined after reduction or oxidation, and the chromium species measured. Cr(VI) and Cr(III) have both been collected on activated charcoal: Cr(VI) as its APDC complex (pH 2.5–3.5) and Cr(III) as the hydroxide form in alkaline solution.12 The presence of active metal on an impregnated activated charcoal surface may greatly enhance the adsorption affinity.13–17 In our previous work, activated charcoal impregnated with Zr (ZrC*) proved to be an efficient and advantageous collector material for Cr(VI) determinations carried out by energy dispersive X-ray fluorescence spectrometry (EDXRF).13
In the work now described, study was made of the adsorption of Cr(III) and Cr(VI) on activated charcoal loaded with Fe (FeC*). On the basis of the results, a separation procedure was developed for the independent determination of Cr(III) and Cr(VI) by graphite furnace atomic absorption spectrometry (GFAAS). The procedure was tested on the Ground Water and Waste Water Pollution Control Check Standard WP-15 spiked with Cr(VI) and waste water samples from a lock manufacturing factory.
2. Experimental
2.1. Apparatus
Measurements were carried out with a Varian SpectrAA 400 atomic absorption spectrometer equipped with a GTA 96 graphite furnace. A hollow cathode lamp filled with neon was used for the measurement of Cr at wavelength 357.9 nm, and a 0.2 nm slit width was selected. Pyrolytic graphite coated tubes (Varian 63-100011-00) were used in all experiments. Unless stated otherwise, the heating cycle was as follows. Dry: 110 °C; ramp 5 s; hold 30 s. Ashing: 1100 °C; ramp 5 s; hold 5 s. Atomisation: 2600 °C; ramp 0.8 s; hold 2 s. The argon flow rate was 3.0 L min−1; the flow was stopped during atomisation.2.2. Standard solutions, reagents and materials
Standard stock solutions (1000 mg L−1 Cr) were prepared for Cr(III) by diluting Titrisol standard (Merck 1.09948) with water, and for Cr(VI) by dissolving 2.8289 g of K2Cr2O7 (Merck 4864) in 1 L of 0.2% HNO3. Iron(III) nitrate (Fe(NO3)3·9H2O, Riedel-de Haën 31233) was used for the preparation of FeC* and was prepared by a published method.18 The amount of modifier metal was 1.10 mmol Fe per g of activated charcoal (AC) (Merck 2186, p.a.). Ground Water and Waste Water Pollution Control Check Standard WP-15 (Spex Industries) was used for the interference studies. All reagents were analytical reagent grade and water (Milli-Q Plus system) was ultrapure grade.2.3. Sample preparation
Synthetic chromium samples of known concentration (10 µg L−1 each of Cr(III) and Cr(VI)) were prepared by diluting known volumes of standard stock solutions with water, and the pH of the sample solution was adjusted to 3.5 ± 0.2 with NaOH or HNO3 solution. The total chromium concentration was measured by GFAAS and for the calibration graph (40–200 pg Cr) standard solutions were prepared from the chromium(III) stock solution by diluting with pure water. Aliquots of 25.0 mL of synthetic chromium samples were transferred to the PE bottles, FeC* (25 mg) was added and the sample solution was carefully stirred and set aside (10 min). The solution was then filtered and unadsorbed chromium (Cr(III)) was diluted in 1% HNO3 and measured by GFAAS. The calibration standard solutions were prepared from the chromium(III) stock solution by dilution with the filtrate of blank solution treated with FeC*. The loaded filter containing adsorbed Cr(VI) was put into another PE bottle and was treated with 25.0 mL of 2.0 M HCl. The solution, together with filter, was then carefully stirred and set aside (20 min) until further filtration. The filtrate containing the Cr(VI) was measured by GFAAS. The calibration standard solutions were prepared from the chromium(VI) stock solution by diluting with the filtrate of a blank solution treated with FeC* and 2.0 M HCl.The WP-15 standard solution was spiked with Cr(VI) and diluted so that concentrations of both Cr(III) and Cr(VI) were 10 μg L−1. Plating samples were diluted (Sample 1 1∶50 and Sample 2 1∶10) with pure water to obtain appropriate concentrations for the separation step. The total chromium concentration of these samples was measured by GFAAS. Aliquots of 25.0 mL were then taken and treated in the same way as synthetic samples.
3. Results and discussion
3.1. Optimisation of GFAAS conditions
GFAAS conditions need to be optimised if accurate and reliable results are to be achieved. Our aim was to optimise the GFAAS so that the two chromium species were atomised equally well in the same furnace program. A variety of difficulties arise in the determination of chromium by GFAAS. The high volatility of Cr compounds leads to significant losses of the metal during thermal pre-treatment in the graphite tube. In some complex matrices, a significant fraction of Cr may be irreversibly retained through the graphite, even at very high atomisation temperatures, by the formation of thermally stable Cr carbides. Background correction has also been a problem. At the resonance wavelength of Cr (357.9 nm), the use of deuterium background correction is questionable because the relative intensity of the emission of the deuterium lamp is too low to ensure an energy balance with the Cr lamp.19 However with our sample matrix background correction proved to be unnecessary.The pyrolysis temperature was studied between 200 and 1700 °C and the shape and size of the signal were good up to 1100 °C for Cr(III) and 1400 °C for Cr(VI). With higher temperatures the signal weakened considerably and broadened. With both chromium species the shape and size of the atomisation signal were good only when the atomisation temperature was between 2500 and 2600 °C; at lower temperatures the signal was broad, low and asymmetrical. Accordingly, 1100 °C was chosen as the pyrolysis temperature and 2600 °C as the atomisation temperature in GFAAS determinations of chromium. Higher pyrolysis temperature would have been possible with the use of chemical modifier, but in this study it was not necessary owing to the low background.
3.2. Separation of Cr(VI) and Cr(III) by FeC* adsorption and GFAAS determinations
In aqueous solution Cr(VI) is present in anionic and Cr(III) in cationic form; also, their adsorption onto untreated activated charcoal differs. Adsorption of Cr(VI) is poor at low pH values, where H2CrO4 is the main species, and in basic solutions where Cr(VI) exists in the form of chromate ion (CrO42−) (Fig. 1). Cr(VI) is better adsorbed in the pH range where chromate ion (HCrO4−) and dichromate ion (Cr2O72−) are in equilibrium, and adsorption is almost quantitative between pH 3 and 4.5. Cr(III) is adsorbed onto unloaded and loaded activated charcoal as hydroxide when the pH of the sample solution increases and the quantitative adsorption (>95%) is achieved at pH range of 8–10. Loading of activated charcoal with Fe improved the adsorption of Cr(VI) significantly; quantitative adsorption (>97%) was then achieved in pH range 3–11 (Fig. 1). At the same time, loading of activated charcoal with Fe hindered the adsorption of Cr(III) completely at low pH values (<4). Iron-loaded activated charcoal is thus well suited for the quantitative separation of Cr(VI) from Cr(III) in pH range 3–4.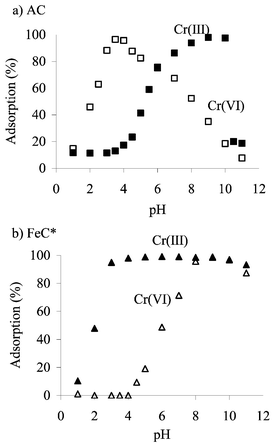 | ||
| Fig. 1 Recoveries of Cr(III) and Cr(VI) onto (a) AC or (b) FeC* as a function of pH of the sample solution (10 µg Cr L−1, V = 25 mL, m(AC) = 25 mg, m(FeC*) = 25 mg, n = 5). | ||
Since the GFAAS method requires liquid samples, the adsorbed Cr(VI) has to be transferred from the loaded FeC* to solution. The most efficient releasing agent proved to be 2.0 M HCl, the recovery of Cr(VI) then being 94%. The volume of HCl releasing solution we used was 25 mL, but this can be decreased if a lower detection limit is required.
The precision for five replicate measurements at the level of 10.0 μg L−1 of Cr(III) and Cr(VI) and the 20.0 µg of the total chromium were calculated to be 2.5%, 5.7% and 3.6% RSD, respectively. The limit of detection based on three times the standard deviation of the blank signal was, for total Cr, 7.1 pg, corresponding to 0.18 µg Cr L−1 in the initial sample volume (25 mL), for Cr(III) 1.8 pg (0.05 µg Cr L−1) and for Cr(VI) 6.8 pg (0.17 µg Cr L−1).
3.3. Interferences
Some matrix elements have been reported to interfere in the determination of chromium by GFAAS. The interference effects were therefore tested with the Ground Water and Waste Water Pollution Control Check Standard WP-15. Besides Cr(III), the standard contains 13 other elements in concentrations varying between 25 and 500 mg L−1. The WP-15 standard solution was spiked with Cr(VI) and diluted so that the concentrations of Cr(III) and Cr(VI) were 10 µg L−1 and the concentrations of the other elements were: Al 50 µg L−1; V 25 µg L−1; As, Be, Co, Cu, Fe, Pb, Mn, Ni, Zn 10 µg L−1; Cd, Se 2.5 µg L−1. Samples were treated as described above. No interferences were observed and recoveries of total Cr, Cr(III) and Cr(VI) were quantitative (Table 1).3.4. Analysis of water samples
The method was applied to the determination of chromium species in wastewater from a lock-manufacturing factory, under optimal experimental conditions. Sample 1 was taken from the process wastewater before purification procedures were applied and sample 2 was from wastewater after purification. To obtain appropriate concentrations of chromium species for the separation steps, samples 1 (10.0 mL) and 2 (50.0 mL) were diluted to 0.500 L with pure water before sample preparation. The measured total chromium and the sum calculated from Cr(III) and Cr(VI) were in good agreement (Table 2). The results show that our new method is well suited for the separation and determination of Cr(III) and Cr(VI) in process wastewater solutions.4. Conclusions
The presence of active metal on the impregnated activated charcoal surface changes the adsorption capacity of the surface towards both Cr(III) and Cr(VI), making their quantitative separation by FeC* possible under appropriate conditions. However, the mechanism of the adsorption of Cr(VI) onto FeC* is still unknown. In the procedure described in this work, total chromium, Cr(III) and Cr(VI) are determined independently, accurately and precisely by GFAAS after quantitative separation of Cr(III) and Cr(VI) from each other by FeC*. The GFAAS procedure was optimised so that total chromium and the two chromium species could be measured under the same conditions. The developed procedure provides good reproducibility, low reagent consumption, simplicity and low detection limits.References
- D. G. Barceloux, Clin. Toxicol., 1999, 37, 173 Search PubMed.
- M. Sperling, Y. Xuefeng and B. Welz, Analyst, 1992, 117, 629 RSC.
- M. Pantsar-Kallio and P. K. G. Manninen, Anal. Chim. Acta, 1996, 318, 335 CrossRef CAS.
- E. Vassileva, K. Hadjiivanov, T. Stoychev and C. Daiev, Analyst, 2000, 125, 693 RSC.
- P. V. Oliveira and E. Oliveira, Fresenius’ J. Anal. Chem., 2001, 371, 909 Search PubMed.
- K. Stein and G. Schwedt, Fresenius’ J. Anal. Chem., 1994, 350, 38 CrossRef CAS.
- D. E. Leyden, R. E. Channel and C. W. Blount, Anal. Chem., 1972, 44, 607 CrossRef CAS.
- C. A. Johnson, Anal. Chim. Acta, 1990, 238, 273 CrossRef CAS.
- T. L. Mullins, Anal. Chim. Acta, 1984, 165, 97 CrossRef CAS.
- D. E. Leyden, K. Goldbach and A. T. Ellis, Anal. Chim. Acta, 1985, 171, 369 CrossRef CAS.
- R. K. Mugo and K. J. Orians, Anal. Chim. Acta, 1993, 271, 1 CrossRef.
- H. A. Van der Sloot, J. Radioanal. Chem., 1977, 37, 727 CAS.
- S. Peräniemi and M. Ahlgrén, Anal. Chim. Acta, 1995, 315, 365 CrossRef.
- S. Peräniemi and M. Ahlgrén, Anal. Chim. Acta, 1995, 302, 89 CrossRef.
- S. Latva, S. Peräniemi and M. Ahlgrén, Anal. Chim. Acta, 2000, 418, 11 CrossRef CAS.
- S. Latva, S. Peräniemi and M. Ahlgrén, Analyst, 1999, 124, 1105 RSC.
- H. Robberrecht and R. van Grieken, Anal. Chem., 1980, 52, 449 CrossRef.
- H. Hashitani, M. Okumura and K. Fujnaga, Fresenius’ J. Anal. Chem., 1987, 326, 540 CAS.
- A. Carlosena, P. López-Mahia, S. Muniategui, E. Fernandéz and D. Prada, J. Anal. At. Spectrom., 1998, 13, 1361 RSC.
| This journal is © The Royal Society of Chemistry 2003 |