Highly efficient acylation of alcohols, amines and thiols under solvent-free and catalyst-free conditions
Brindaban C. Ranu*, Suvendu S. Dey and Alakananda Hajra
Department of Organic Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Calcutta, 700 032, India. E-mail: ocbcr@iacs.res.in; Fax: 91-33-4732805
First published on 14th January 2003
Abstract
A general, simple, efficient, cost-effective and green procedure for acylation of alcohols, amines and thiols has been developed by treatment with acid anhydride or acid chloride at 80–85 °C under solvent and catalyst-free conditions.
Green ContextGreen chemistry can be considered as a set of reductions and for chemical processes a reduction in the complexity of that process is fundamentally important. This means reducing the use of auxiliaries (solvent, reagent, catalyst, etc.) and reducing the waste. Here we see an example of green chemistry reductions applied to the widely used acylation of functional groups. The methodology described here employs substrates only, with no solvent or catalyst and the product can be removed directly from the process by distillation.JHC |
Introduction
The acylation of functional groups, especially hydroxyl and amino groups is one of the most basic and frequently used transformations in organic synthesis as it provides a useful and efficient protection protocol in a multistep synthetic process.1 Moreover, this reaction has a biomimetic importance as acylation of xenobiotics is a metabolic pathway that increases lipophilicity. Acylation is usually carried out by treatment of an alcohol or amine with acetyl chloride or acetic anhydride in the presence of an acid or a base catalyst in a suitable organic solvent, although acetic anhydride is the most commonly used being less toxic.1 The most efficient base catalysts are 4-(dimethylamino)pyridine (DMAP),2a and phosphines,2b,c and the powerful acid catalysts employed include CoCl2,3a TaCl5,3b Cu(OTf)2,3c Me3SiOTf,3d Sc(OTf)3,3e,f In(OTf)3,3g Bi(OTf)3,3h LiClO43i and yttria-zirconica based Lewis acid.3j The solvents commonly chosen for these reactions are methylene chloride, acetonitrile and tetrahydrofuran. Thus, apart from some operational disadvantages like moisture-sensitivity and high cost of many of these catalysts these procedures using toxic metal derivatives and chlorinated hydrocarbons as solvents also do not satisfy the requirements of green synthesis. Thus, a need for a practical, efficient and greener alternative for this important transformation prompted us to disclose here a simple procedure for acylation without any solvent and catalyst (Scheme 1). Although there are a few examples of solvent-free acylation in the presence of an acid or a base particularly for amides and imides4 to the best of our knowledge, no generalized and systematic study for acylation of alcohols, amines and thiols has been made under solvent- and catalyst-free conditions with a view to developing a practical methodology.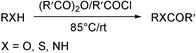 | ||
| Scheme 1 | ||
Results and discussion
In a typical general procedure, a neat mixture of an alcohol and an acid anhydride was heated at 80–85 °C without any solvent and catalyst under nitrogen for a certain period of time as required to complete the reaction. The acetate was distilled out directly from the reaction vessel under reduced pressure.A wide range of structurally diverse and functionalised alcohols underwent acylation by this procedure to provide the corresponding acetates in excellent yields. The results are reported in Table 1. As evident from the results, this procedure is uniformly effective for acylation of primary (including saturated, allylic, propargylic and benzylic) and secondary alcohols, phenols and diols. Acylations of hindered secondary alcohol (entry 34) and tertiary alcohol (entry 36) which are often considered difficult, are also accomplished in reasonably good yields. Carbohydrate molecules such as glucose, galactose and sucrose undergo complete acylation of all the hydroxyl groups (entries 39–41). This procedure is also applicable for efficient acylation of thiols, amines and oximes (entries 48–54). Although most of the reactions were carried out with acetic anhydride other acid anhydrides such as propionic and benzoic anhydride are also equally effective (entries 5, 6, 15, 16, 30 and 31). Acid chlorides can also be used for acylation under similar experimental conditions at room temperature (entries 7, 8, 17, 18, 32 and 33). Most significantly, tolerance to several acid-sensitive functionalities such as acetal (entry 23), methylenedioxy (entry 22), carboxylic ester (entries 37 and 45) shows the superiority of this methodology over many existing procedures using strong Lewis acids.
| Entry | Substrate | Anhydride/acid chloride | Time/h | Yield of acetatea (%) | Entry | Substrate | Anhydride/acid chloride | Time/h | Yield of acetatea (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Yields refer to those of pure isolated products characterized by spetral data (IR, 1H, 13C NMR).b Reaction carried out at room temperature. | |||||||||
| 1 | CH3(CH2)5OH | (MeCO)2O | 1.5 | 92 | 28 | Cyclopentanol | (MeCO)2O | 2 | 90 |
| 2 | CH3OCH2CH2OH | (MeCO)2O | 1.5 | 94 | 29 | Cyclohexanol | (MeCO)2O | 2 | 92 |
| 3 | CH3CH2OCH2CH2OH | (MeCO)2O | 1.5 | 93 | 30 | Cyclohexanol | (EtCO)2O | 2 | 90 |
| 4 | PhCH2OH | (MeCO)2O | 2 | 95 | 31 | Cyclohexanol | (PhCO)2O | 1.5 | 89 |
| 5 | PhCH2OH | (EtCO)2O | 1.5 | 88 | 32 | Cyclohexanol | MeCOCl | 0.75 | 89b |
| 6 | PhCH2OH | (PhCO)2O | 1.5 | 90 | 33 | Cyclohexanol | EtCOCl | 0.75 | 88b |
| 7 | PhCH2OH | MeCOCl | 0.5 | 91b | 34 | 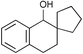 | (MeCO)2O | 2 | 84 |
| 8 | PhCH2OH | EtCOCl | 0.5 | 92b | 35 | (−)-Menthol | (MeCO)2O | 2 | 89 |
| 9 | PhCH2CH2OH | (MeCO)2O | 1.75 | 91 | 36 | t-Butanol | (MeCO)2O | 3 | 78 |
| 10 | PhCH2CH2OH | (PhCO)2O | 1.5 | 91 | 37 | Diethyl L-tartarate | (MeCO)2O | 2.5 | 76 |
| 11 |  | (MeCO)2O | 1.25 | 89 | 38 | Glycerol | (MeCO)2O | 2.5 | 70 |
| 12 | HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2OH CCH2OH | (MeCO)2O | 1.5 | 91 | 39 | D-Glucose | (MeCO)2O | 2.5 | 70 |
| 13 |  | (MeCO)2O | 1.25 | 87 | 40 | D-Galactose | (MeCO)2O | 2.5 | 75 |
| 14 | PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2OH CHCH2OH | (MeCO)2O | 1.5 | 92 | 41 | Sucrose | (MeCO)2O | 2.5 | 73 |
| 15 | PhCHCHCH2OH | (EtCO)2O | 1.5 | 85 | 42 | Phenol | (MeCO)2O | 1.5 | 80 |
| 16 | PhCHCHCH2OH | (PhCO)2O | 2 | 86b | 43 | 4-(OMe)C6H4OH | (MeCO)2O | 1.5 | 82 |
| 17 | PhCHCHCH2OH | MeCOCl | 0.75 | 90b | 44 | 4-(NO2)C6H4OH | (MeCO)2O | 1.5 | 84 |
| 18 | PhCHCHCH2OH | EtCOCl | 0.75 | 91 | 45 | Ethyl salicylate | (MeCO)2O | 1.5 | 89 |
| 19 | 4-(O-Allyl)C6H4CH2OH | (MeCO)2O | 2 | 91 | 46 |  | (MeCO)2O | 2 | 74 |
| 20 | 4-(OBz)C6H4CH2OH | (MeCO)2O | 2 | 89 | 47 | Catechol | (MeCO)2O | 1.5 | 75 |
| 21 | 4-(NO2)C6H4CH2OH | (MeCO)2O | 2 | 88 | 48 | PhSH | (MeCO)2O | 1.5 | 82 |
| 22 | Piperonyl alcohol | (MeCO)2O | 2 | 90 | 49 | PhSH | (EtCO)2O | 1 | 90 |
| 23 | 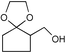 | (MeCO)2O | 1.75 | 92 | 50 | PhSH | MeCOCl | 0.5 | 93b |
| 24 | 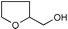 | (MeCO)2O | 1.75 | 74 | 51 |  | (MeCO)2O | 1.5 | 85 |
| 25 | 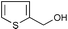 | (MeCO)2O | 1.5 | 79 | 52 | PhNH2 | (MeCO)2O | 0.5 | 93 |
| 26 | CH3CH2CH(OH)CH3 | (MeCO)2O | 2 | 90 | 53 | Cyclohexyl amine | (MeCO)2O | 1 | 79 |
| 27 | PhCH(OH)CH3 | (MeCO)2O | 2 | 91 | 54 | 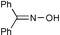 | (MeCO)2O | 1.5 | 80 |
The acylations are, in general reasonably fast and clean. No side product has been isolated in any reaction. The products obtained by direct distillation from the reaction vessel are of high purity and do not require further purification. As a test case, a couple of reactions are scaled up to the extent of a batch with 10 g of alcohol (amount not optimised) without any difficulty avoiding use of any organic solvent in any step.
Conclusion
The present solvent-free and catalyst-free procedure provides a powerful and versatile acylation method for alcohols, thiols, amines and oximes. This method is endowed with several unique merits, namely, simplicity in operation, mild reaction conditions tolerable to acid sensitive functionalities, wide applicability and cost-efficiency. Moreover, this protocol introduces a practical and viable green technology of solvent-free and catalyst-free reactions.5 Further investigations to broaden the scope of this technology are in progress.Experimental
General
All alcohols, thiols and amines are commercial materials and are distilled before use. Acetic, propionic and benzoic anhydrides and acid chlorides were freshly distilled and stored over anhydrous potassium carbonate.General experimental procedure for acylation
A neat mixture of an alcohol (thiol or amine) (5 mmol) and an acid anhydride (or acid chloride) (6 mmol) was gently heated with stirring at 80–85 °C (oil-bath) without any solvent and catalyst under nitrogen for a certain period of time as required for a complete reaction (monitored by TLC). The resulting acetate was distilled out directly from the reaction vessel under reduced pressure. Alternatively, for a lower scale reaction if distillation is not convenient, the product was extracted with diethyl ether. The ether extract was washed with aqueous sodium bicarbonate solution followed by brine and evaporated to leave the crude product which was purified by short column chromatography over silica gel.The acetates are all well known compounds and are easily identified by their IR, 1H NMR and 13C NMR spectral data in comparison to those reported.
Acknowledgements
This investigation has enjoyed the financial support from CSIR, New Delhi [Grant No. 01(1739)/02]. S. S. D. and A. H. are also thankful to CSIR for their fellowships.References
- (a) T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 3rd edn., 1999, p. 150 Search PubMed; (b) R. C. Larock, Comprehensive Organic Transformations, Wiley-VCH, New York, 2nd edn., 1999, p. 1955 Search PubMed.
- (a) W. Steglich and G. Hofle, Angew. Chem., Int. Ed. Engl., 1969, 8, 981 CrossRef; (b) E. Vedejs, N. S. Bennet, L. M. Conn, S. T. Diver, M. Gingras, S. Lin, P. M. Oliver and M. J. Peterson, J. Org. Chem., 1993, 58, 7286 CrossRef CAS; (c) E. Vedejs and J. A. Mackay, Org. Lett., 2001, 3, 535 CrossRef CAS.
- (a) J. Iqbal and R. R. Srivastava, J. Org. Chem., 1992, 57, 2001 CrossRef CAS; (b) S. Chandrasekhar, T. Ramchander and M. Takhi, Tetrahedron Lett., 1998, 39, 3263 CrossRef CAS; (c) P. Sarvanan and V. K. Singh, Tetrahedron Lett., 1999, 40, 2611 CrossRef CAS; (d) P. A. Procopiou, S. P. D. Baugh, S. S. Flack and G. G. A. Inglis, J. Org. Chem., 1998, 63,, 2342 CrossRef CAS; (e) K. Ishihara, M. Kubota, H. Kurihara and H. Yamamoto, J. Org. Chem., 1996, 61, 4560 CrossRef CAS; (f) J.-C. Lee, C.-A. Tai and S.-C. Hung, Tetrahedron Lett., 2002, 43, 851 CrossRef CAS; (g) K. K. Chauhan, C. Frost, G. I. Love and W. Waite, Synlett, 1999, 1743 CAS; (h) A. Orita, C. Tanahashi, A. Kakuda and J. Otera, J. Org. Chem., 2001, 66, 8926 CrossRef CAS; (i) Y. Nakae, I. Kusaki and T. Sato, Synlett,, 2001, 1584 CrossRef CAS; (j) P. Kumar, R. K. Pandey, M. S. Bodas and M. K. Dongare, Synlett, 2001, 206 CAS.
- (a) D. Davidson and H. Skovronek, J. Am. Chem. Soc., 1958, 80, 376 CrossRef CAS; (b) C. D. Hurd and A. G. Prapas, J .Org. Chem,, 1959, 24, 388 CrossRef CAS.
- M. Joselink, R. S. Varma, S. Polanc and M. Kocevar, Chem. Commun., 2001, 1716 RSC.
| This journal is © The Royal Society of Chemistry 2003 |