Exhaustive hydrodechlorination of chlorinated aromatic environmental pollutants to alicyclic compounds
Amal Ghattas, Raed Abu-Reziq, David Avnir* and Jochanan Blum*
Department of Organic Chemistry, The Hebrew University, Jerusalem, 91904, Israel. E-mail: david@chem.ch.huji.ac.il and jblum@chem.ch.huji.ac.il
First published on 6th January 2003
Abstract
A combined Pd–Rh catalyst obtained by co-entrapment of metallic palladium and soluble [Rh(cod)Cl]2 within a silica sol–gel matrix exhaustively detoxifies chlorinated aromatic pollutants not only by total removal of the chlorine, but also by the total reduction of the aromatic moieties to alicyclic rings. The list of chloroarenes which have been successfully destroyed by the catalysts includes most of the major pollutants which are of public concern, namely chlorinated dibenzo[b,e]dioxin and dibenzofuran, PCBs, DDT, Hexachlorophene and 2,4,5-T. The reactions take place under the relatively mild conditions of 80–100 °C under 27 atm H2. The immobilised catalyst is leach proof in a variety of solvents and can be reused in several runs. The high activity of the combined Pd–Rh system is attributed to synergism between the two different metal atoms.
Green ContextWhile green chemistry seeks to avoid the formation of pollutants through highly efficient clean processes and the production of benign products, we will always have pollutants to deal with. Chlorinated organic pollutants represent one of the major targets for treatment. It is important that in carrying out such treatments we do not add to the overall environmental burden through unreasonable use of additional resource (including energy) or through the production of secondary pollutants. Here we see described a new hetergeneous catalyst for the hydrodechlorination of chlorinated aromatic pollutants. The reactions take place under quite mild conditions (minimum energy demands) and the precious metal catalyst is reusable (minimum resource demands).JHC |
Introduction
Public concern over chlorinated organic pollutants1 has fueled many efforts to develop efficient and clean destruction methods of these chemicals. Several processes have been developed towards this goal, most of which are reductive procedures that remove the chlorine but leave the aromatic hydrocarbons unaffected.2 However, as benzene and some of its derivatives, as well as polycyclic aromatic hydrocarbons (PAH) are by themselves hazardous carcinogenic substances, it seems imperative to develop detoxification processes that destroy the aromatic moieties as well. So far only a few studies in this direction, leading from chlorinated benzene derivatives to cyclohexanes, have been announced.3,4 However, to the best of our knowledge, no general efficient catalyst that transfers all major pollutants—polychlorinated dibenzo[b,e]dioxin (PCDDs), dibenzofuran (PCDFs), and biphenyl (PCBs), as well as 1,1′-(2,2,2-trichloroethylidene)bis(4-chlorobenzene) (DDT), 2,2′-methylenebis(3,4,6-trichlorophenol) (Hexachlorophene) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T)—directly to HCl and totally reduced aromatics, has been reported. Here we describe such a catalyst, namely a sol–gel-entrapped combined Pd–[Rh(cod)Cl]2 system. This recyclable system has already been shown to promote the hydrogenation of aromatic C–C bonds,5 as well as the direct transformation of benzylic CO to CH2 moieties.6Results and discussion
Our initial hydrogenation experiments of some simple chloroarenes in the presence of the sol–gel entrapped Pd-[Rh(cod)Cl]2 catalyst are listed in Table 1. Under our experimental conditions (80–100 °C and 27 atm H2) the Pd–Rh catalyst system is stable and recyclable. Several of the reactions listed in Table 1 were repeated 3–4 times with the same catalyst without significant loss in its activity. The same catalyst has also been used occasionally for the hydrogenation of different substrates. It is notable that the HCl eliminated in these reactions does not accumulate in the sol–gel matrix. The chlorine content of the HF-treated immobilized catalyst prior to the catalytic process and after it was found to be identical. Although the hydrogenation process both eliminates the ring-bound chlorine atom and saturates the aromatic C–C bonds, simple alkyl chlorides are not affected. Therefore, it is possible to carry out the reactions in 1,2-dichloroethane in which many chloroarenes are highly soluble. The hydrodechlorination process can however, be performed equally well in chlorine-free alkanes. Other common solvents such as methanol or THF have also been used, but the reaction proceeded at a lower rate (see Table 1, entries 4 and 5). In water, in which the chloroarenes are usually only sparingly soluble the reaction proceeded even slower. Although steric hindrance seems to have some effect on the combined dechlorination and aromatic ring hydrogenation, the various vicinal di-, tri- and tetrachlorobenzenes, listed in Table 1 gave, under our standard conditions, solely cyclohexane, free of chlorinated side products. Only the reaction mixture of pentachlorobenzene consisted after 6 h of the fully hydrogenated cyclohexane together with 10% of tetrachlorobenzene which gradually disappeared upon extension of the reaction time. Hexachlorobenzene, however did not react at all. We presume that the unreactivity of hexachlorobenzene and the slow reaction of tetrachlorobenzene are associated with steric effects. 1-Chloronaphthalene (entry 11), was found to form a mixture of tetralin and cis- and trans-decalin. Although linear chloroalkanes are not dechlorinated, entries 12 and 13 indicate that chlorine atoms at the benzylic position undergo hydrogenolysis similar to aryl chlorides. It is remarkable that under the conditions of Table 1, the chlorobenzenes do not yield detectable amounts of chlorine-free reaction intermediates (i.e., benzene, cyclohexadienes or cyclohexene). However, the hydrogenation of 3- and 4-chlorophenol forms, apart from cyclohexanol, considerable amounts of cyclohexanone which can be regarded as the keto tautomer of 1-cyclohexenol. Interestingly, while the hydrogenation of chlorophenols in 1,2-dichloroethane yields, apart from the carbinol and the ketone also cyclohexane, in water the oxygen functions do not undergo hydrogenolysis.| Entry | Substrate | Solvent | Reaction time/h | Product (yield, %)b |
|---|---|---|---|---|
| a Reaction conditions: 2 mmol substrate, ceramic catalyst containing 2 × 10−2 [Rh(cod)Cl]2 and 0.1 mmol Pd, 5 ml solvent, 100 °C, 27 atm. H2., stirring rate 150 rpm.b The yield is an average of at least two runs that did not differ by more than 5%. The missing percentage is usually accounted for by unreacted starting material.c Mixture of hexanes containing ∼85% of n-hexane. | ||||
| 1 | C6H5Cl | 1,2-C2H4Cl2 | 3 | C6H12 (87) |
| 2 | 1,2-C6H4Cl2 | 1,2-C2H4Cl2 | 3 | C6H12 (91) |
| 3 | 1,2-C6H4Cl2 | Hexanec | 3 | C6H12 (100) |
| 4 | 1,2-C6H4Cl2 | MeOH | 3 | C6H12 (70) |
| 5 | 1,2-C6H4Cl2 | THF | 3 | C6H12 (20) |
| 6 | 1,3-C6H4Cl2 | 1,2-C2H4Cl2 | 3 | C6H12 (78) |
| 7 | 1,4-C6H4Cl2 | Hexanec | 3 | C6H12 (100) |
| 8 | 1,2,4-C6H3Cl3 | 1,2-C2H4Cl2 | 6 | C6H12 (32) |
| 9 | 1,2,3,4-C6H2Cl4 | 1,2-C2H4Cl2 | 6 | C6H12 (43) |
| 10 | 1,2,3,4,5-C6HCl5 | 1,2-C2H4Cl2 | 6 | C6H12 (63), 1,2,4,5-C6H2Cl4 (10) |
| 11 | 1-C10H7Cl | Hexanec | 3 | C10H12 (71), c-C10H18 (11), t-C10H18 (17) |
| 12 | C6H5CH2Cl | 1,2-C2H4Cl2 | 6 | C6H11CH3 (95) |
| 13 | C6H5CCl3 | Hexanec | 6 | C6H11CH3 (96) |
| 14 | 3-ClC6H4OH | H2O | 6 | C6H11OH (44), C6H10O (26) |
| 15 | 4-ClC6H4OH | 1,2-C2H4Cl2 | 4 | C6H12 (37), C6H11OH (25), C6H10O (28) |
| 16 | 4-ClC6H4OH | H2O | 4 | C6H11OH (49), C6H10O (21) |
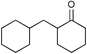
Next we studied the hydrogenation of some major pollutants in the presence of the sol–gel entrapped Pd–[Rh(cod)Cl]2 catalyst. The results are summarised in Table 2. The Table indicates that in the banned insecticide DDT7 both the ring-bound, as well as the chlorine atoms at the homobenzylic position undergo hydrogenolysis and 1,1-dicyclohexylethane is formed in a high yield (Table 2, entry 1). The former bactericide and antiseptic agent Hexcachlorophene which turned out to be neurotoxic8 yields mainly dicyclohexylmethane, but in analogy to the chlorophenols listed in Table 1, gives also a ketone, 2-cyclohexylmethyl(cyclohexanone)9 (entry 2). The infamous plant growth regulator and herbecide, 2,4,5-T10 proved to give under our standard conditions only chloroacetyl chloride and cyclohexane (entry 3). As a representative example for the polychlorinated biphenyls (PCBs)11,12 we hydrogenated a commercial mixture of 2,2′,3,3′,4,4′-, 2,2′,3,4,4′5- and 2,2′4,4′,5,5′-hexachloro-1,1′-biphenyl (PCB-128, PCB-138 and PCB-153, respectively). In this experiment (Table 2, entry 4) chlorine-free bicyclohexyl was obtained in quantitative yield. Using 1,2,4-trichlorodibenzo[b,e][1,4]dioxin13 and chlorinated dibenzofuran14 as model compounds, for polychlorinated dibenzodioxins (PCDDs) and polychlorinated dibenzofurans (PCDFs) the high efficiency of our catalyst was demonstrated again through exhaustive hydrogenation of these compounds (entries 5 and 6). While the dioxin derivative yielded almost entirely pure dodecahydrodibenzo[b,e][1,4]dioxin,15 the dodecahydrodibenzofuran16 was contaminated with up to 18% of a mixture of chlorine- and oxygen-free non-aromatic hydrocarbons.
| Entry | Substrate | Solvent | Product | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions as in Table 1. Time 24 h. The hexane used contained ∼85% n-hexane.b Average of at least 2 runs. Except for entry 6, the missing percentage is accounted for by unreacted starting material.c The amount of chloroacetyl chloride in the reaction mixture was not determined.d Mixed with 16% of 2,3,8-trichlorodibenzofuran.e Contaminated with 19% of chlorine- and oxygen-free non-aromatic hydrocarbons. | ||||
| 1 | 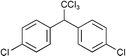 | Hexane | 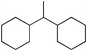 | 90 |
| 2 | 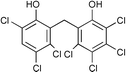 | 1,2-C2H4Cl2 | 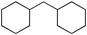 | 58 |
 | 32 | |||
| 3 | 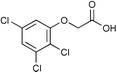 | 1,2-C2H4Cl2 |  | 75c |
| 4 | 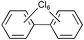 | Hexane | 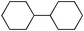 | 99 |
| 5 | 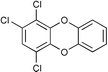 | 1,2-C2H4Cl2 | 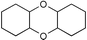 | 93 |
| 6 | 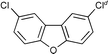 d d | 1,2-C2H4Cl2 | 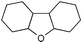 | 81e |
| 7 | 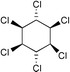 | 1,2-C2H4Cl2 | 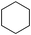 | 18 |
As noted above the refractory behaviour of aliphatic chloroalkanes toward hydrogenation, under our experimental conditions, enabled us to carry out the reactions in 1,2-dichloroethane. Some cyclic chlorohydrocarbons could however be dechlorinated. Of particular interest is the hydrodechlorination of the hexachlorocyclohexanes. The γ-isomer, Lindane, which is sold worldwide under many different trademarks as an insecticide is reduced slowly (18% within 24 h) under the conditions of Table 2 (entry 7). Chlorinated intermediates were not detected in the reaction mixture.
In contrast to some other catalytic hydrogenations of chloroarenes (e.g., see references 2e and 2s), the liberated hydrogen chloride seems neither to interfere nor to enhance the progress of the process. However, attempts to trap the HCl gas by running the reaction in an autoclave beaded with Ba(OH)2 resulted in a decrease of the reaction rate.
The pronounced catalytic activity of the combined Pd–Rh system is attributed to synergism that may exist between the sol–gel entrapped palladium and the metal of the encaged [Rh(cod)Cl]2 complex. While the combined catalyst system promoted the conversion of 1,2-dichlorobenzene in 1,2-C2H4Cl2 to give 91% of cyclohexane within 3 h (Table 1, entry 2), the reaction with rhodium-free entrapped palladium afforded only 30% of cyclohexane and the heterogenized [Rh(cod)Cl]2 gave under the same conditions 19% of the alicyclic product.
Although this study is concerned with the hydrodehalogenation of chloroarenes, it is notable that fluoroarenes undergo similar defluorination albeit at somewhat lower rates. Under the conditions of Table 1, fluorobenzene is converted into cyclohexane in 50% yield within 3 h (as compared with the yield of 87% obtained from chlorobenzene under the same conditions). Bromoarenes are hydrodehalogenated to give chiefly halogen-free aromatic products. Under the aforementioned conditions, bromobenzene gives after 3 h 80% of benzene and 18% of cyclohexane. We attribute the partial poisoning of the catalyst to the formation of HBr. A similar phenomenon has already been observed in a previous study on hydrogenation of haloarenes by the sol–gel entrapped ion pair RhCl3–Aliquat 336.4
In conclusion, we propose that this general catalytic process by the immobilised Pd–Rh catalyst may also serve as a facile way for disposal of other unwanted chloroarenes that form unfavoured benzene derivatives by the conventional hydrodechlorination methods.
Experimental
General procedure for hydrodechlorination of aromatic chlorine compounds
Typically, a glass lined autoclave, equipped with a mechanical stirrer, a sampler and a temperature controller was charged with the chlorinated substrate (2 mmol), the appropriate solvent (5 ml) and a sol–gel entrapped catalyst5 prepared from Na2PdCl4 (32 mg, 0.11 mmol), NaBH4 (22 mg), H2N(CH2)3Si(OCH3)3 (0.27 ml), [Rh(cod)Cl]2 (10 mg, 0.02 mmol) and Si(OCH3)4 (1.7 ml). The Pd and Rh loading in the heterogenized catalyst so formed, was 1.45 and 0.52% respectively, the diameters of the Pd particles were 2–4 nm, and the N2-Langmiur surface area of the ceramic materials was 730 m2 g−1 with a narrow pore size distribution centered around 15 Å. The sealed autoclave was flushed three times with hydrogen and pressurized to 27 atm. The reaction mixture was stirred at a rate of 150 rpm at 100 °C for the required length of time. After cooling to room temperature the autoclave was opened, the catalyst separated by filtration, washed and sonicated with dichloromethane, dried and reused in a second run. The filtrate was concentrated and the reaction products separated, if necessary by GC, and analyzed by comparing their IR, NMR and MS spectra with those of authentic samples.Acknowledgements
We thank Roy Jackson and the staff of the Centre of Green Chemistry at Monash University, Melbourne for most helpful discussions, and the Israel Science Foundation, as well as the Ministry of Science, Culture and Sport for financial support of this study.References
- See, e.g. B. Hileman, J. R. Long and E. M. Kirschner, Chem. Eng. News, 1994, 72(47), 12 Search PubMed.
- For some recent publications see e.g., (a) F. Murena and E. Schioppa, Appl. Catal. B: Environ., 2000, 27, 257 CrossRef CAS; (b) C. Schœth, S. Disser, F. Schüth and M. Reinhard, Appl. Catal. B: Environ., 2000, 28, 147 CrossRef; (c) C. Menini, C. Park, E.-J. Shin, G. Tavoularis and M. A. Keane, Catal. Today, 2000, 62, 355 CrossRef CAS; (d) M. A. Aramendía, V. Borau, I. M. Gracía, C. Jiménez, A. Marinas, J. M. Marinas and F. J. Urbano, C.R. Acad. Sci. Paris, Sér. IIc, Chem./Chem., 2000, 3, 465 Search PubMed; (e) F. Murena, E. Schioppa and F. Gioia, Environ. Sci. Technol., 2000, 34, 4382 CrossRef CAS; (f) N. Lingaiah, M. A. Uddin, A. Muto, T. Iwamoto, Y. Sakata and Y. Kusano, J. Mol. Catal. A: Chem., 2000, 161, 157 CrossRef CAS; (g) C. M. King, R. B. King, N. K. Bhattacharyya and M. G. Newton, J. Organomet. Chem., 2000, 600, 63 CrossRef CAS; (h) P. Tundo, A. Perosa and S. S. Zinovyev, Appl. Catal. B: Environ., 2001, 32, 1L CrossRef; (i) Y. Cesteros, P. Salagre, F. Medina, J. E. Sueiras, D. Tichit and B. Coq, Appl. Catal. B: Environ., 2001, 32, 25 CrossRef CAS; (j) I. Yamanaka, K. Nishikawa and K. Otsuka, Chem. Lett., 2001, 368 CrossRef CAS; (k) E. Schioppa, F. Murena and F. Gioia, Ind. Eng. Chem. Res., 2001, 40, 2011 CrossRef CAS; Y. Shindler, Y. Matatov-Meytal and M. Sheintuch, Ind. Eng. Chem. Res., 2001, 165, 9; (l) G. Del Angeles and J. L. Benitez, J. Mol. Catal. A: Chem., 2001, 165, 9 CrossRef; (m) Y. A. Serguchev and Y. V. Belokopytov, Kinet. Catal., 2001, 42, 174 Search PubMed; (n) V. I. Simagina and I. V. Stoyanova, Mendeleev Commun., 2001, 38 CrossRef; (o) B. H. Lipshutz, T. Tomioka and K. Sato, Synlett., 2001, 970 CrossRef CAS; (p) F. Murena and F. Gioia, Appl. Catal. B: Environ., 2002, 38, 39 CrossRef CAS; (q) Y. Ukisu and T. Miyadera, Chemosphere, 2002, 46, 507 CrossRef CAS; (r) R. Gopinath, K. Narasimha Rao, P. S. Sai Prasad, S. S. Madhavendra, S. Narayanan and G. Vivekanandan, J. Mol. Catal. A: Chem., 2002, 181, 215 CrossRef CAS; (s) M. A. Aramendía, V. Boráu, I. M. Gracía, C. Jiménez, F. Lafont, A. Marinas, J. M. Marinas and F. J. Urbano, J. Mol. Catal. A: Chem., 2002, 184, 237 CrossRef CAS; (t) C. Desmarets, S. Kuhl, R. Schneider and Y. Fort, Organometallics, 2002, 21, 1554 CrossRef CAS.
- See, e.g. (a) S. T. Srinivas, P. S. Sai Prasad, S. S. Madhavendra and P. Kanta Rao, in Recent Advantages in Basic and Applied Aspects of Industrial Catalysis, Stud. Surf. Sci. Catal., ed. T. S. R. Prasada Rao and G. Murali Dhar, Elsevier, Amsterdam, 1998, vol. 113, p. 835 Search PubMed; (b) Y. Cesteros, P. Salagre, F. Medina and J. E. Sueiras, Catal. Lett., 2000, 67, 147 Search PubMed; (c) W. D. Marshall, A. Kubátová, A. J. M. Lagadec, D. J. Miller and S. D. Hawthorne, Green Chem., 2002, 4, 17 RSC.
- J. Blum, A. Rosenfeld, F. Gelman, H. Schumann and D. Avnir, J. Mol. Catal. A: Chem., 1999, 146, 117 CrossRef CAS.
- R. Abu-Reziq, D. Avnir, I. Miloslavski, H. Schumann and J. Blum, J. Mol. Catal. A: Chem., 2002, 185, 177 CrossRef.
- R. Abu-Reziq, D. Avnir and J. Blum, J. Mol. Catal. A: Chem., 2002, 187, 277 CrossRef CAS.
- See: R. L. Metcalf, in Ullmann′s Encyclopedia of Industrial Chemistry, ed. B. Elver, S. Hawkins, M. Ravenscroft and G. Schulz, VCH, Weinheim, 5th edn., 1989, vol. 14, pp. 278–280 Search PubMed.
- Status of Pesticides in Reregistration and Special Review, Office of Pesticide Programs, U.S. Environmental Protection Agency, Washington, D.C., 1992 Search PubMed.
- J. von Braun and O. Bayer, Ber. Dtsch. Chem. Ges, 1926, 59, 2317 Search PubMed.
- M. L. Leng, in Ullmann′s Encyclopedia of Industrial Chemistry, ed. W. Gerhartz, Y. S. Yamamoto, F. T. Campbell, R. Pfefferkorn and J. F. Rounsville, VCH, Weinheim, 5th edn., 1986, vol. 7, pp. 9–16 and references therein Search PubMed.
- See, e.g. U. Beck, in Ullmann′s Encyclopedia of Industrial Chemistry, ed. W. Gerhartz, Y. S. Yamamoto, F. T. Campbell, R. Pfefferkorn and J. F. Rounsville, VCH, Weinheim, 5th edn., 1986, vol. 6, pp. 347–355 Search PubMed.
- S. H. Safe, in Kirk-Othmer Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz and M. Howe-Grant, Wiley-Interscience, New York, 4th edn., 1993, vol. 6, pp. 127–139 Search PubMed.
- A. E. Pohland and G. C. Yang, J. Agric. Food Chem, 1972, 20, 1093 CrossRef CAS.
- The chlorinated dibenzofuran prepared by the procedure of A. P. Gray, V. M. Dipinto and I. J. Solomon, J. Org. Chem., 1976, 41, 2428 Search PubMed was a mixture of 84% of 2,8-di- and 16% of 2,3,8-trichlorodibenzofuran.
- The IR, 1H NMR and GCMS spectra were practically identical with those of a sample of perhydrodibenzo[b,e][1,4]dioxin prepared from cyclohexen oxide and PdCl2 according to J. A. Scheben and I. L. Mador, US Pat., 3,480,632, to National Distillers and Chemical Corp., 25 Nov. 1969 (Chem. Abstr. 1970, 72, 43694h).
- The compound proved identical with a sample obtained by exhaustive hydrogenation of dibenzofuran: J. I. Jones and A. S. Lindsey, J. Chem. Soc., 1950, 1836 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |