New food antioxidant additive based on hydrolysis products of lactose
A. Abbadi*a, K. F. Gotliebb, J. B. M. Meibergc and H. Van Bekkumd
aAraChem Contract Research & Custom Synthesis, P.O. Box 2021, 6020, AA Budel, The Netherlands. E-mail: a.abbadi@arachem.nl
bStoepveldsingel 119, 9403, SM Assen, The Netherlands
cGlucona B.V, P.O. Box 15, 9640, AA Veendam, The Netherlands
dDelft University of Technology, Laboratory of Organic Chemistry and Catalysis, Julianalaan 136, 2628, BL Delft, The Netherlands
First published on 16th January 2003
Abstract
The hydrolysis products of lactose, D-glucose and D-galactose, were oxidized in two consecutive steps using appropriate bimetallic catalysts (Bi–Pd, Bi–Pt on charcoal) to the corresponding 2-ketoaldonic acids. The latter were further processed to provide two stereoisomers of L-ascorbic acid (vitamin C), D-ascorbic acid (D-lyxoascorbic acid) and erythorbic acid (D-araboascorbic acid). The antioxidant activity of these two isomers as well as that of the equimolar mixture thereof was evaluated by measuring their reducing activity against DPPH (2,2-diphenyl-1-picrylhydrazyl) and their scavenging activity of O2− species in the presence of NBT (Nitro Blue Tetrazolium). The compounds and their equimolar mixture were found to exhibit the same antioxidant activity as L-ascorbic acid suggesting that they may be used as cheap alternatives to L-ascorbic acid as food antioxidant additives when the vitamin C activity is not required.
Green ContextL-Ascorbic acid is now a large volume chemical product with applications in vitamin preparations and as an antioxidant in food. The current industrial manufacturing process is multi-step and the high cost of the product limits the scope of its applications. Here it is shown that the equimolar mixture of D-glucose and D-galactose derived stereoisomers of L-ascorbic acid is a promising alternative antioxidant where vitamin C is not required. The substitute mixture is cheaper and can be prepared directly by the hydrolysis of lactose. Thus a useful environmentally benign product can be prepared by an efficient simple route in a way that enables its more widespread application.JHC |
Introduction
The industrial production of L-ascorbic acid has now reached a volume of ca. 60000 t/a. The industrial process is a five-step synthesis starting from D-glucose and involves a regioselective biocatalytic oxidation of sorbitol. The overall yield of L-ascorbic acid from D-glucose is presently ca. 60% and its current price is about 14€/kg. About one-third of the total production is used for vitamin preparations in the pharmaceutical industry. The remainder is mainly applied as an antioxidant additive to food and feed to enhance product quality and stability.1The most significant characteristic of L-ascorbic acid is its oxidation to dehydro-L-ascorbic acid with which it forms a reversible redox system (Scheme 1). L-Ascorbic acid is a strong reducing agent due to its enediol structure. The oxidation of L-ascorbic acid proceeds in two steps via the intermediate radical anion, semidehydro-L-ascorbic acid, to provide dehydro-L-ascorbic acid. Semidehydro-L-ascorbic acid is a strong acid with a pKa of 0.45 and acts as a good radical scavenger.2
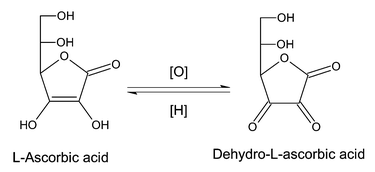 | ||
| Scheme 1 L-Ascorbic acid/dehydro-L-ascorbic acid reversible redox system. | ||
The vitamin C activity of L-ascorbic acid is related to the enediol lactone group, the R-configuration for the C-4 chiral center, the presence of at least two carbon substituents on C-4 and the L-configuration for the C-5 hydroxyl group. The primary C-6 hydroxyl group has a minor impact on the biological activity. Of the other three diastereomers of L-ascorbic acid only erythorbic acid (D-araboascorbic acid) shows some antiscorbutic activity, ca. 5% of that of vitamin C.3
In applications where the vitamin C activity is not important, erythorbic acid, possessing the required enediol structure to act as an antioxidant, is often used as a cheaper alternative to L-ascorbic acid. Some of the applications of the antioxidant and reducing properties of L-ascorbic acid are:1
• inhibition of nitrosamine formation in cured meat products
• protection toward enzymatic browning of processed fruits and vegetables
• improvement of flour baking qualities of dough and the appearance of baked goods
• retardation of oxidative rancidity in fats and oils
• increased clarity of wine and beer
• synergetic activity in applications with other antioxidants (e.g., tocopherols)
• as a developing agent in photography, etc…
The potential use of L-ascorbic acid in agriculture (improvement of seed germination, ozone protection, harvesting aid) is limited by its price. In this work we have applied the present state of art in the selective catalytic oxidation of carbohydrates4 to prepare a cheaper antioxidant and reducing agent mixture from the less expensive and readily available and also renewable raw material lactose (Scheme 2).
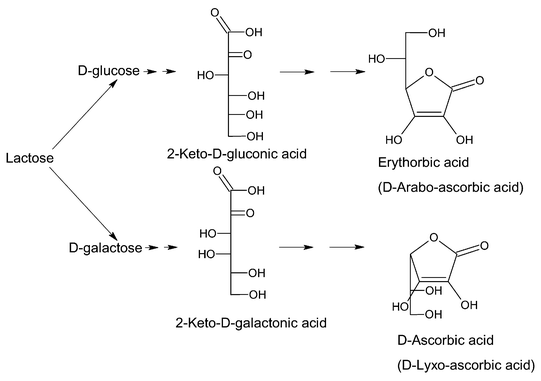 | ||
| Scheme 2 Synthetic pathway for the formation of the two stereoisomers of L-ascorbic acid, erythorbic acid (D-araboascorbic acid) and D-ascorbic acid (D-lyxoascorbic acid) from lactose. | ||
Lactose, or milk sugar, is among the most abundant of carbohydrates and its principal source is (sweet) whey, the main by-product in cheese and casein production in the dairy industry.5 Due to the size of the dairy industry in the EU, only a part of the whey is further processed to lactose. The EU production of lactose is currently 250000 t/a. One third of the lactose produced is used in the pharmaceutical industry while the remainder is consumed in the manufacture of various foods and infant nutrition products. Much effort has been made to develop new processes and products based on lactose to enlarge its total market.6–16 Among the commercial products derived from lactose are lactitol (ca. 6000 t/a), lactulose (ca. 20.000 t/a) and Ca–lactobionate (ca. 200 t/a) obtained by hydrogenation, isomerization and oxidation, respectively. This work is also aimed toward an enlargement of the lactose market by developing new products based on this raw material. Upon hydrolysis of lactose an equimolar mixture of D-glucose and D-galactose is obtained. These two monosaccharides were submitted to consecutive known catalytic reactions to provide a cheaper antioxidant and reducing agent based on the L-ascorbic acid stereoisomers, D-ascorbic acid (D-lyxoascorbic acid) and erythorbic acid (D-araboascorbic acid). The antioxidant/reducing properties of these stereoisomers as well as their 1∶1 mixture have been compared with those of L-ascorbic acid.
Results and discussion
The aim of this work was to develop new cheap food antioxidant additives by further processing of the D-glucose/D-galactose equimolar mixture obtained upon hydrolysis of lactose. The resulting product after processing is a 1∶1 mixture of stereoisomers of L-ascorbic acid with antioxidant/reducing properties. Prior to studying the preparation of this antioxidant mixture directly from lactose, we have undertaken the synthesis of each stereoisomer of L-ascorbic acid separately by starting from the corresponding monosaccharide. Furthermore, we have evaluated the antioxidant activity of these stereoisomers as well as the activity of their equimolar mixture. Their performance was compared to that of L-ascorbic acid. Two methods were applied for the evaluation of the antioxidant activity:i. measurement of the antioxidant reducing activity against DPPH (2,2-diphenyl-1-picrylhydrazyl).17
ii. measurement of the antioxidant scavenging activity of O2− species in the presence of the dye NBT (Nitro Blue Tetrazolium).18,19
Synthesis of D-glucose and D-galactose derived stereoisomers of L-ascorbic acid
The monosaccharide (aldose) was first oxidized to the corresponding aldonic acid using a Bi–Pd on carbon catalyst with oxygen acting as the oxidant. The oxidation reaction was carried out under alkaline conditions (pH = 9) as described in the literature.20,21 The pH was kept constant during the oxidation by addition of a solution of 2 M NaOH using a Dosimat. Under these reaction conditions a total conversion of the aldose to aldonic acid was achieved with a selectivity higher than 98%.The sodium aldonate obtained was further submitted to oxygen-catalyzed oxidation over a Bi–Pt on carbon catalyst to provide the corresponding sodium 2-ketoaldonate. The oxidation reaction was conducted as described previously22 without pH control with a conversion and selectivity higher than 98%.
The sodium 2-ketoaldonate was then converted to its corresponding acid form by treatment with a cation exchange resin and the free acid was esterified with methanol in the presence of 5 wt% dry HCl as the catalyst. The methyl ester was finally treated with sodium methoxide to provide the corresponding isomer of L-ascorbic acid by cyclo-transesterification. Details in connection with the synthetic procedures are reported in the Experimental section.
Evaluation of the antioxidant activity
We have applied DPPH and NBT as reagents for the evaluation of the antioxidant activity of the D-galactose and the D-glucose derived stereoisomers of L-ascorbic acid as well as the equimolar mixture of these two compounds. The activity measured was compared to that of L-ascorbic acid. The results are reported below.| Antioxidant | Concentration/M | Reduced DPPH (%) |
|---|---|---|
| a The listed concentrations are the sum of the concentrations of the two stereoisomers. | ||
| L-Ascorbic acid | 1 × 10−5 | 16.6 |
| 5 × 10−5 | 94.8 | |
| 1 × 10−4 | 98.6 | |
| Erythorbic acid | 1 × 10−5 | 17.2 |
| (D-araboascorbic acid) | 5 × 10−5 | 93.4 |
| 1 × 10−4 | 99.4 | |
| D-Ascorbic acid | 1 × 10−5 | 17.7 |
| (D-lyxoascorbic acid) | 5 × 10−5 | 94.1 |
| 1 × 10−4 | 99.4 | |
| 1∶1 mixturea of erythorbic | 1 × 10−5 | 17.1 |
| acid/D-ascorbic acid | 5 × 10−5 | 93.7 |
| 1 × 10−4 | 99.2 | |
We have applied this method to compare the O2− scavenging activity of L-ascorbic acid with its stereoisomers obtained from D-glucose, D-galactose as well as their equimolar mixture. The sample mixture was left for 30 min at 25 °C whereby the enzymatic reaction was stopped by addition of CuCl2 (see Experimental section). The results are summarized in Table 2.
| Antioxidant | Concentration | Formazan formed (%) |
|---|---|---|
| a The listed concentrations are the sum of the concentrations of the two stereoisomers.b No formazan is formed. | ||
| L-Ascorbic acid | 1 × 10−6 | 93.3 |
| 1 × 10−5 | 35.6 | |
| 1 × 10−4 | n.fb | |
| Erythorbic acid | 1 × 10−6 | 94.6 |
| (D-araboascorbic acid) | 1 × 10−5 | 40.7 |
| 1 × 10−4 | n.f | |
| D-Ascorbic acid | 1 × 10−6 | 95 |
| (D-lyxoascorbic acid) | 1 × 10−5 | 35.6 |
| 1 × 10−4 | n.f | |
| 1∶1 mixturea of erythorbic | 1 × 10−6 | 95.5 |
| acid/D-ascorbic acid | 1 × 10−5 | 38 |
| 1 × 10−4 | n.f | |
From these results we can conclude that the O2− scavenging activity of the two stereoisomers studied as well as that of their equimolar mixture is comparable to that of L-ascorbic acid. The formation of Blue Formazan was totally inhibited by a concentration of 10−4 M of the antioxidant in the reaction mixture, suggesting that all O2− species generated by the xanthine oxidase/xanthine system were scavenged prior to any reaction with NBT.
Conclusion
The D-glucose and D-galactose derived stereoisomers of L-ascorbic acid as well as their equimolar mixture exhibit the same activity as L-ascorbic acid toward the reduction of DPPH and the scavenging of the in situ-generated active O2− species.The equimolar mixture of D-glucose and D-galactose derived stereoisomers of L-ascorbic acid is a promising and inexpensive antioxidant that may be used as a cheaper alternative to L-ascorbic acid when the vitamin C activity is not required.
The optimization of the different steps of the process to prepare this antioxidant mixture directly from the D-glucose/D-galactose equimolar mixture, readily obtained by hydrolysis of lactose, is in progress to evaluate the feasibility and the economy of this new food antioxidant additive.
Experimental
Materials
L-Ascorbic acid, DPPH, NBT, xanthine, the disodium salt of EDTA, 5% Pt/C and bismuth(III) nitrate were purchased from Aldrich. Xanthine oxidase and a 5% solution of albumin (BSA) were obtained from Sigma. Solvents, all of analytical grade, D-glucose, D-galactose and 5% Pd/C were purchased from Acros Organics. All the reagents were used without further purification.Synthesis of D-glucose and D-galactose derived stereoisomers of L-ascorbic acid
The apparatus used, the analysis procedures, as well as the start-up of the oxidation reactions have already been described.22,23 The preparation of the 5% Bi/5% Pd/C catalyst has been reported in the literature.20,21i. Oxidation of the aldose to sodium D-aldonate
13C NMR (75 MHz, D2O): δ 181.1, 73.1, 72.9, 71.6, 71.3, 64.9.
13C NMR (75 MHz, D2O): δ 180.2, 75.6, 74.1, 72.7, 72.5, 64.1.
ii. Oxidation of sodium aldonate to sodium 2-ketoaldonate
The 5% Bi/5% Pt/C catalyst was prepared in the same manner as the 5% Bi/5% Pd/C catalyst. The start-up procedure for the heterogeneous selective catalytic oxidation of aldose was applied for the preparation of the sodium 2-ketoaldonate derivatives. The oxidation reaction was carried out at 50 °C but without pH control22 using 1 g of the catalyst for 5 g of sodium aldonate. After 2.5 h the consumption of oxygen ceased and a sample was taken from the reaction mixture and analyzed by HPLC. A total conversion of sodium aldonate to the corresponding 2-ketoaldonic acid with a selectivity >98% was observed. The oxidation reaction was then stopped, the catalyst was removed by filtration and the filtrate was concentrated to provide sodium 2-keto-D-adonate on standing at 0 °C.13C NMR (75 MHz, D2O): δ 173.6, 98.2, 72.6, 72.1, 67.5, 64.2.
13C NMR (75 MHz, D2O): δ 176.2, 96.7, 71.5, 70.8, 65.9, 63.6.
iii. Synthesis of 2-keto-aldonic acid methyl ester
Sodium 2-keto-D-aldonate, obtained from the previous oxidation, was dissolved in deionized water, treated with a cation exchange resin (type Dowex in the H+ form) and freeze-dried to offer the corresponding 2-keto-D-aldonic acid.8 g of 2-keto-D-aldonic acid was suspended in 100 mL of methanol containing 5 wt% dry HCl and the mixture was refluxed under nitrogen for 4 h. The reaction mixture was then cooled to room temperature under nitrogen and concentrated in vacuum. The syrup obtained was allowed to crystallize overnight at 0 °C to provide the methyl ester of 2-keto-D-aldonic acid. The crude product was further purified by crystallization from methanol.
13C NMR (75 MHz, D2O): δ 172.2, 98.3, 72.6, 72.0, 67.3, 64.2, 54.9.
13C NMR (75 MHz, D2O): δ 171.9, 98.3, 73.1, 72.5, 67.6, 64.3, 55.3.
iv. Cyclization of the methyl ester of 2-ketoaldonic acid
5 g of the methyl ester was dissolved under nitrogen in 50 mL of methanol and the temperature of the solution was set at 68 °C. To this solution was added 1 equivalent of sodium methoxide whereupon an instantaneous reaction took place and a precipitate of the sodium salt of the corresponding stereoisomer of L-ascorbic acid was formed. The reaction was allowed to proceed for 5 min then the reaction mixture was cooled in an ice-bath. The suspension was treated with a previously dried cation exchange resin to prepare the free form of the L-ascorbic acid stereoisomer. After 15 min, the cation exchange resin was removed by filtration and the clear solution was evaporated in vacuum. The residue obtained was crystallized at 0 °C from small amount of methanol to provide the desired antioxidant.13C NMR (75 MHz, D2O): δ 172.1, 153.6, 115.8, 79.4, 70.9, 64.0.
1H NMR (300 MHz, D2O): δ 4.5 (d, 1H), 4.0 (m, 1H), 3.7 (q, 2H).
13C NMR(75 MHz, D2O): δ 174.9, 157.2, 119.4, 77.8, 70.5, 63.6.
1H NMR (300 MHz, D2O): δ 4.9 (d, 1H), 4.0 (m, 1H), 3.7 (t, 2H).
Measurement of the antioxidant reducing activity against DPPH
Weighed amounts of the test compound was dissolved in 5 mL of DMF and diluted to 100 mL with ethanol to give 10−5, 10−4, 10−3 and 10−2 M solutions. To 5 mL of a 2 × 10−4 M solution of DPPH in ethanol was added 4 mL of ethanol and 1 mL of the test compound solution. After standing for 20 min at 25 °C the absorbance of the sample was measured at 517 nm. The difference in absorbance from the control (no test compound added) was taken as a measure of the reducing activity.Measurement of the antioxidant scavenging activity of O2− species in the presence of Nitro Blue Tetrazolium (NBT)
To 50 mL of sodium carbonate buffer solution (pH = 10.2) was added 22.81 mg of xanthine (3 × l0−4 M), 31.33 mg of NBT (7.5 × 10−5 M), 55.83 mg of Na2H2EDTA (3 × l0−4 M) and a weighed amount of the test compound to prepare sample solutions with concentrations of 10−6, 10−5, 10−4 and 10−3 M. Further, in a separate flask, a mixture was prepared containing a 1% (v/v) xanthine oxidase suspension from buttermilk (>0.5 units/mg protein) in an aqueous solution of 0.15% (v/v) albumin from bovine serum (BSA).To 5 mL of the buffered mixture was added 0.16 mL of the solution containing xanthine oxidase. The mixture was incubated at 25 °C for 30 min. The enzymatic reaction was stopped by addition of 1 mL of a 6 mM aqueous solution of CuCl2 and the absorbance of the reaction mixture was measured at 560 nm (the maximum absorbancy of Blue Formazan). The decrease in the absorbance relative to the control was taken as a measure for the scavenging activity.
Acknowledgement
The authors wish to express their gratitude to Glucona B.V., Veendam, The Netherlands, for financial support.References
- B. Oster and U. Fechtel, Ullmann′s Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 1990, vol. A27, p. 547 Search PubMed.
- B. H. J. Bielski, in Ascorbic Acid: Chemistry, Metabolism, and Uses, ed. P.A. Seib and P. M. Tolbert, American Chemical Society, Washington, D.C., 1982, p. 81 Search PubMed.
- F. Smith, in Advances in Carbohydrate Chemistry, ed. W. W. Pigman, M. L. Wolfrom and S. Peat, Academic Press, New York, 1946, vol. 2, p. 79 Search PubMed.
- A. Abbadi and H. van Bekkum, in Carbohydrates as Organic Raw Materials III, ed. H. van Bekkum, H. Röper and A. G. J. Voragen, VCH/CRF, New York, 1996, p. 37 Search PubMed.
- E. Timmermans, in Carbohydrates as Organic Raw Materials III, ed H. van Bekkum, H. R. Röper and A. G. J. Voragen, VCH/CRF, New York, 1996, p. 93 Search PubMed.
- CCA Biochem B. V., Eur. Pat., 039 981, 1981.
- C. A. Lobry de Bruin and W. Alberda van Ekenstein, Recl. Trav. Chim. Pays-Bas, 1895, 14, 203 Search PubMed.
- H. E. J. Hendriks, B. F. M. Kuster and G. B. Marin, Carbohydr. Res., 1990, 204, 121 CrossRef CAS.
- J. B. M. Meiberg, P. M. Bruinenberg and B. Sloots, Eur. Pat., 384 534 1990.
- G. de Wit, J. J. de Vlieger, A. C. Kock-van Dalen, R. Heus, R. Laroy. A. J. van Hengstum, A. P. G. Kieboom and H. van Bekkum, Carbohydr. Res., 1981, 125, 91.
- H. Walton, US Pat., 2 752 334 1956.
- T. J. Williams, N. R. Plessas and I. J. Goldstein, Carbohydr. Res., 1978, 67, C1 CrossRef.
- H. E. J. Hendriks, B. F. M. Kuster and G. B. Marin, Carbohydr. Res., 1991, 214, 71 CrossRef CAS.
- R. van den Berg, J. A. Peters and H. van Bekkum, Carbohydr. Res., 1995, 267, 65 CrossRef CAS.
- A. Abbadi, K. F. Gotlieb, J. B. M Meiberg and H. van Bekkum, J. Appl. Catal., 1997, 156, 105 Search PubMed.
- A. Abbadi, K. F. Gotlieb, J. B. M Meiberg and H. van Bekkum, Green Chem., 1999, 231 RSC.
- M. S. Blois, Nature, 1958, 181, 1199 CAS.
- C. Beauchamp and I. Fridovich, Anal. Biochem., 1971, 44, 276 CrossRef CAS.
- M. Nishikimi, Biochem. Biophys. Res. Commun., 1975, 63, 463 CrossRef CAS.
- M. Besson, P. Gallezot, F. Lahmer, G. Fleche and P. Fuertes, in Catalysis of Organic reactions, ed. J. R. Kosak and T. A. Johnson, Marcel Dekker, New York, 1994, p. 169 Search PubMed.
- B. M. Despeyroux, K. Deller and E. Peldszus, in New Developments in Selective Oxidation (Stud. Surf. Sci. Catal., Vol. 59), ed. G. Centi and F. Trifiro, Elsevier, Amsterdam, 1990, p. 159 Search PubMed.
- A. Abbadi and H. Van Bekkum, J. Appl. Catal., 1995, 124, 409 Search PubMed.
- A. Abbadi, M. Makkee, W. Visscher, J. A. R. Van Veen and H. Van Bekkum, J. Carbohydr. Chem., 1993, 12, 573 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |