A novel and simple method for the silylation of alcohols in DMSO–hexane without a catalyst
Tsutomu Watahiki, Masaya Matsuzaki and Takeshi Oriyama*
Department of Environmental Sciences, Faculty of Science, Ibaraki University, 2-1-1 Bunkyo, Mito, 310-8512, Japan. E-mail: tor@mx.ibaraki.ac.jp
First published on 16th January 2003
Abstract
Reaction of alcohols with tert-butyldimethylsilyl chloride in DMSO–hexane provides the corresponding tert-butyldimethylsilyl ethers in high yields under mild reaction conditions. This convenient reaction proceeds very smoothly at room temperature without a catalyst. Additionally, alcohols having an allylic alcohol, cyclopropane, and tetrahydrofuran moiety underwent chemoselective silylation to give the TBS ethers in good yields with no undesired reactions.
Green ContextSilyl ethers are a very popular class of compounds in organic chemistry largely due to the versatility of the function as a protecting group. They are normally prepared by the reaction of an alcohol with a trialkylsilyl halide using at least a stoichiometric quantity of base. The desired product is then accompanied by a salt, which has to be separated. Thus the standard procedure employs an auxiliary, involves a separation stage and produces a waste product. In this paper this conventional thinking is challenged—do we need a base? By screening various solvents the authors find that reaction will occur without base and that with DMSO hexane in particular, quantitative formation of the silyl ether could be achieved.JHC |
Introduction
Silyl ethers are a most popular and promising protecting group of hydroxy functions in synthetic organic chemistry, and various types of silyl ethers have been developed so far.1 Silyl ethers are very easy to introduce and to remove, and have general stability for most non-acidic reagents and high solubility in non-polar solvents. Silyl ethers are commonly obtained by the reaction of the parent alcohols with the corresponding trialkylsilyl halide in the presence of stoichiometric amounts of a base, such as imidazole,2 4-dimethylaminopyridine,3N,N-diisopropylethylamine,4 and so on.5 However, these base-catalyzed silylation methods have a serious disadvantage, since careful extraction and filtration processes are required to remove annoying ammonium salts.6 Very recently, we have reported novel catalyst-less reactions: direct conversion of alcohol tetrahydropyranyl ethers or alcohol triphenylmethyl ethers into the corresponding acetates by the reaction with acetyl bromide.7 These transformations have some outstanding green-chemical features. From the viewpoint of green chemistry, a catalyst-less reaction is very significant and attractive in synthetic organic chemistry. However, efficient silylation of alcohols without a catalyst has been scarcely reported.On the other hand, it has been recently reported that spontaneous aldol and Michael reactions of enoxytrimethylsilanes proceed smoothly in dipolar aprotic solvents, such as dimethyl sulfoxide (DMSO).8 This reaction presumably proceeds via an activation of the enoxysilane by coordination of the DMSO oxygen atom to the silicon atom. Therefore, we envisioned that if a trialkylsilyl chloride was activated by DMSO, reaction of alcohols with the trialkylsilyl chloride would take place in DMSO to yield the corresponding trialkylsilyl ethers in high yields. In this communication, we report a simple and efficient method for the silylation of alcohols with trialkylsilyl chloride in DMSO with no catalyst.
Results and discussion
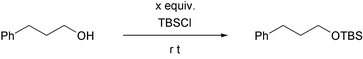
To begin with, we examined the solvent effects for the silylation of 3-phenyl-1-propanol (Table 1). The reaction of 3-phenyl-1-propanol with 1.2 equiv. of tert-butyldimethylsilyl chloride in N,N-dimethylformamide (DMF) for 0.5 h afforded the corresponding tert-butyldimethylsilyl (TBS) ether in 37% yield (run 1). On the other hand, when the reaction was carried out in DMSO, the reaction took place very smoothly and the corresponding TBS ether was obtained in 75% isolated yield (run 2). In the case of using other solvents such as CH2Cl2, THF, hexane, AcOEt, and MeCN, the corresponding TBS ethers were obtained in lower yields (runs 3–7). From these results, it could be presumed that DMSO strongly activated the silicon atom of trialkylsilyl chloride.
Next, we investigated the equiv. of the reagent and the loading of DMSO (Table 2). On increasing the equiv. of tert-butyldimethylsilyl chloride, the yields of TBS ethers were greatly improved (runs 1–3). However, treatment of alcohol with 2 equiv. of silyl chloride during 18 h gave the TBS ether in lower yield (run 4). This is owing to deprotection of the produced silyl ether with in situ-generated HCl. Therefore, to prevent the deprotection, the reaction was performed with 3 equiv. of DMSO in hexane. After completion of the reaction, the corresponding TBS ether was obtained in 94% yield (run 5). In spite of the extension of reaction time for 18 h, no deprotection of once formed TBS ether was observed under the reaction conditions (run 6).
Representative and successful examples for the synthesis of various alcohol TBS ethers are collected in Table 3. As well as primary alcohols, secondary alcohols were readily transformed into the corresponding TBS ethers in high yields (runs 3–5). In the case of tertiary alcohol and phenol, the yields of TBS ethers were rather lower (runs 6 and 7).
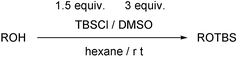
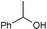
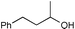
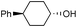

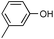
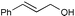
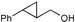
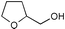



Additionally, alcohols having an allylic alcohol, cyclopropane, and tetrahydrofuran moiety underwent chemoselective silylation to give the TBS ethers in good yields with no undesired reactions (runs 8–10). In the presence of other protecting groups of the hydroxy function, silylation of alcohols also proceeded chemoselectively with these protecting groups unaffected (runs 11–13).
Finally, in order to establish the generality of the benign reaction, we attempted the silylation of 3-phenyl-1-propanol with various trialkylsilyl chlorides (Table 4). By using tert-butyldiphenylsilyl or triphenylsilyl chloride, which are a sterically hindered silyl moiety, the corresponding tert-butyldiphenylsilyl (TBDPS) or triphenylsilyl (TPS) ethers were similarly formed in high yields (runs 2 and 3). In contrast, the reaction of alcohol with triisopropylsilyl chloride gave the corresponding triisopropylsilyl (TIPS) ether in only 23% yield (run 4).

In conclusion, we have accomplished a new method for the synthesis of silyl ethers from alcohols in DMSO–hexane. This reaction proceeds very smoothly at room temperature and the desired silylated products were obtained in high yields under mild reaction conditions. By this method, neither a base nor an acid catalyst is necessary to complete the reaction. Therefore, this catalyst-less method is broadly applicable to the silylation of alcohols having base- or acid- sensitive functional groups. In addition to its significance as a green-chemical method, this method serves also potential utility in the syntheses of complex natural products. Further research addressing the scope and elucidation of the reaction mechanism is under way in our laboratory.
Experimental
Typical procedure for the silylation of alcohol in DMSO–hexane
To a solution of 3-phenyl-1-propanol (40.9 mg, 0.3 mmol) in hexane (0.5 ml) was added dimethyl sulfoxide (64 μl, 0.9 mmol) and tert-butyldimethylsilyl chloride (67.8 mg, 0.45 mmol) at room temperature under an argon atmosphere. The resultant mixture was stirred for 3 h at room temperature and quenched with water. The organic materials were extracted with Et2O and dried over anhydrous magnesium sulfate. The solvent was evaporated and tert-butyldimethyl(3-phenylpropoxy)silane (70.7 mg, 94%) was isolated by thin-layer chromatography on silica gel (ether∶hexane = 1∶20). The product gave satisfactory NMR and IR spectra.Spectroscopic data
Acknowledgements
T. W. thanks the Sasakawa Scientific Research Grant from The Japan Science Society for generous financial support.References
- T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1999 Search PubMed; P. J. Kocienski, Protective Groups, Georg Thieme Verlag, New York, 3rd edn., 1994 Search PubMed.
- E. J. Corey and A. Venkateswarlu, J. Am. Chem. Soc., 1972, 94, 6190 CrossRef CAS.
- S. K. Chaudhary and O. Hernandez, Tetrahedron Lett., 1979, 99 CrossRef CAS.
- L. Lombardo, Tetrahedron Lett., 1984, 25, 227 CrossRef CAS.
- G. A. Olah, B. G. B. Gupta, S. C. Narang and R. Malhotra, J. Org. Chem., 1979, 44, 4272 CrossRef CAS; G. R. Martinez, P. A. Grieco, E. Williams, K. Kanai and C. V. Srinivasan, J. Am. Chem. Soc., 1982, 104, 1436 CrossRef CAS; J. M. Aizpurua and C. Palomo, Tetrahedron Lett., 1985, 26, 475 CrossRef CAS; S. Kim and H. Chang, Bull. Chem. Soc. Jpn., 1985, 58, 3669 CAS; B. A. D’Sa, D. Mcleod and J. G. Verkade, J. Org. Chem., 1997, 62, 5057 CrossRef CAS.
- Allylsilane and silyl enol ether could also silylate alcohols under the influence of a catalytic amount of p-toluenesulfonic acid, trifluoromethanesulfonic acid or iodine. These methods do not need a work-up process, such as an extraction: T. Morita, Y. Okamoto and H. Sakurai, Tetrahedron Lett., 1980, 21, 835 Search PubMed; T. Veysoglu and L. A. Mitscher, Tetrahedron Lett., 1981, 22, 1299 CrossRef CAS; G. A. Olah, A. Husain, B. G. B. Gupta, G. F. Salem and S. C. Narang, J. Org. Chem., 1981, 46, 5212 CrossRef CAS; A. Hosomi and H. Sakurai, Chem. Lett., 1981, 85 CrossRef CAS; T. Suzuki, T. Watahiki and T. Oriyama, Tetrahedron Lett., 2000, 41, 8903 CAS.
- T. Oriyama, K. Kobayashi, T. Suzuki and M. Oda, Green Chem., 2002, 4, 30 RSC; K. Kobayashi, T. Watahiki and T. Oriyama, Synthesis Search PubMed , in press.
- Y. Génisson and L. Gorrichon, Tetrahedron Lett., 2000, 41, 4881 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |