Recent developments on the epoxidation of alkenes using hydrogen peroxide as an oxidant
G. Grigoropouloua, J. H. Clark*a and J. A. Elingsb
aCentre for Clean Technology, Chemistry Department, University of York, U.K YO10 5DD. E-mail: gclog@york.ac.uk
bQuest International, Ashford, Kent, UK TN24 0LT
First published on 20th December 2002
Abstract
This paper reviews recent developments on the epoxidation of alkenes using hydrogen peroxide as an environmentally friendly oxidant. The activation of hydrogen peroxide takes place in the presence of a homogeneous metal catalyst such as tungten, manganese, iron and rhenium or through addition of stoichiometric amounts of auxiliaries, which convert hydrogen peroxide to a more active oxidant. Hydrogen peroxide has also been used with solids catalysts such as mixed oxides, redox zeolites, layered materials and supported metal catalysts. Heterogeneous catalysts, however, suffer generally from lower activity and stability compared to the homogeneous catalysts.
Green ContextThe epoxidation of alkenes is one of the key steps in functionalising hydrocarbons, as well as rapidly building functionality into a range of molecules, often with excellent control over selectivity. This review covers the development of clean epoxidation chemistry, in particular focussing on those methods which involve hydrogen peroxide, which is arguably the ‘greenest’ terminal oxidant in the continued absence of a general direct method to directly utilise oxygen in this transformation.JWV |
Introduction
Epoxides are highly useful intermediates for the manufacture of a range of important commercial products.1 Their selective synthesis is a subject of considerable academic and industrial interest. Olefin epoxidation is one of the main routes, which leads to the production of epoxides on both a laboratory and industrial scale.2The use of oxygen, peroxides and peracids for direct oxidation of alkenes is the main method for industrial applications. Ethylene is commercially epoxidised by vapour-phase oxidation with oxygen or air using a supported silver catalyst.3 However, this catalytic method is not efficient for alkenes with allylic C–H bonds due to oxidation at this position. Propylene oxide is produced by the metal catalysed liquid phase oxidation of propylene using organic hydroperoxides produced by hydrocarbon autoxidation.4 Homogeneous5 MoVI and heterogeneous6 TiIV are used efficiently as catalysts and tert-butyl hydroperoxide or ethylbenzene hydroperoxide as the oxidants. Even though, the method has been widely used for the production of epoxides, it suffers from the formation of the alcohol co-product (tert-butanol or 1-phenylethanol).
Epoxidation of substituted alkenes widely used in the fine chemicals industry can be successfully achieved by using stoichiometric amounts of peracids such as peracetic acid and m-chlorobenzoic acid.7 However, the employment of peracids is not a clean method as equivalent amounts of acid waste are produced. The safety issues associated with handling peracids is also a matter for concern.
There is a strong need for the development of new epoxidation methods which employ safer oxidants and produce little waste. The employment of hydrogen peroxide is an attractive option both on environmental and economic grounds. It is cheap, readily available and gives water as the only by-product. Many catalytic systems based on different metals such as tungsten, manganese, rhenium and titanium have been reported for the epoxidation of a wide range of alkenes using hydrogen peroxide. Furthermore, immobilisation of active homogeneous catalysts on solid supports has attracted a lot of interest as heterogeneous catalysts are easily separated from the product mixture and can possibly be recycled. Here, we critically review recent literature on the synthesis of epoxides using hydrogen peroxide as the terminal oxidant.
Heteropolyoxometalates
Transition metal substituted polyoxometalates have attracted considerable interest as oxidatively stable catalysts, 8–10 however, chlorinated solvents are normally required in order to obtain high reaction rates and yields of epoxides.An improved catalytic system was developed by Noyori and co-workers11,12 containing sodium tungstate dihydrate, aminomethylphosphonic acid and a quaternary ammonium salt hydrogen sulfate with 30% hydrogen peroxide in the absence of solvent or alternatively in toluene (Fig. 1). Lipophilic quaternary ammonium hydrogen sulfate significantly enhances the yield to epoxide. This catalytic system is effective for the epoxidation of terminal and cyclic olefins. The reaction, however, fails to produce acid sensitive epoxides due to their hydrolytic decomposition under the acidic conditions.
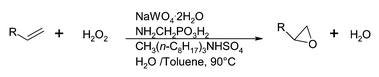 | ||
| Fig. 1 Epoxidation of alkenes by phosphotungstate catalysts under PTC conditions. | ||
A quaternary ammonium cetylpyridinium heteropolyoxotungstate π-[C5H5NC16H33]3[PO4(WO3)4] based catalyst with special solubility characteristics has recently been reported13 for the catalysis of the epoxidation of 1-hexene and cyclohexene at high selectivities (>87%). The catalyst becomes soluble in aqueous hydrogen peroxide–toluene due to the in situ formation of π-[C5H5NC16H33]3{PO4[W(O)2(O2)]4}. At the end of the reaction in the absence of hydrogen peroxide, the catalyst precipitates out, facilitating its recovery from the reaction mixture. The catalyst is stable and can be recycled without loss of activity.
The catalytic efficiency of a series of ‘sandwich’ type transition metal substituted polyoxometalates of the general formula NaxM2Zn3W19O68 (M = Ru, Mn, Zn, Pd, Pt, Co, Fe, Rh) has been examined in the epoxidation of different alkenes using hydrogen peroxide as a terminal oxidant under biphasic conditions in the presence of tricaprylammonium chloride as a phase transfer catalyst.14 [WZnMnII2(ZnW9O34)2]12− efficiently catalyses the epoxidation of a wide range of alkenes such as acyclic and cyclic alkenes, and alkenols with high chemoselectivity and stereospecificity. The other transition metal substituted polyoxometalates degrade in the presence of aqueous hydrogen peroxide and the active species formed efficiently catalyses the epoxidation of primary alkenols only. Therefore, those catalysts suffer from a significant loss of their activity and low recyclability due to their degradation.
Even though most of the polyoxometalates catalysed epoxidations are limited to alkenes containing one double bond, novel manganese containing heteropolyanions have been efficiently applied to the regioselective epoxidation of (R)-(+)-limonene.15 A series of manganese(II) substituted polyoxometalates such as [(MnII(H2O)3)2(WO2)2(BiW9O33)2]10−, [(MnII(H2O))3(SbW9O33)2]12− and [(MnII(H2O)3)2(MnII(H2O)2)2(TeW9O33)2]8− have been synthesised and gave high yields of 1,2-limonene oxide at ambient temperature. Furthermore, remarkably high turnovers (>2500 after 24 h) at higher ratios of substrate–oxidant–catalyst (5000∶10000∶1) have been achieved. The use of dichloromethane as a solvent is critical for the reaction, as the catalysts are not soluble enough in other solvents such as acetonitrile, acetone and ethanol.
Epoxidation of terpenes such as α-pinene, 3-carene and linalool has been effectively catalysed by a heterogeneous tungsten based catalyst16 which was obtained by the exchange of a commercial, macroreticular Amberlite IRA-900 with the Venturello anion {PO4[WO(O2)2]4}3−. The catalyst is very active and highly selective (>90%) towards the epoxide in the oxidation of limonene (Fig. 2), unsaturated C10 alcohols and their esters. However, the epoxidation of acid sensitive terpene epoxides such as α-pinene oxide was only achieved under homogeneous conditions at 83% conversion and 92% selectivity by adding a preformed Venturello complex [(C8H17)3NCH3]3[PW4O24] to the reaction mixture. In the latter case, benzene or toluene has been used as solvent instead of halogenated solvent which is normally used under Venturello conditions.17
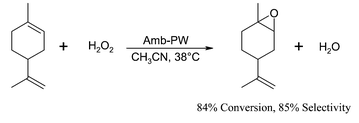 | ||
| Fig. 2 Epoxidation of limonene by Amberlite-supported tungsten catalyst. | ||
The immobilisation of tungsten and phosphotungsten (PW) catalytic species on both organic resins such as Amberlite IRA-900 and hybrid silica materials has been examined in the epoxidation of bulky olefins.18 Silica supports modified with quaternary ammonium functionalities provide a stable charged support where PW species are supported by electrostatic interactions. Alternatively, PW species were covalently supported on a phosphoramide modified silica. The ion exchange of the preformed Venturello complex {PO4[WO(O2)2]4}3− on the Amberlite support leads to efficient catalysts. However, in situ formation of peroxo–W species bound to the immobilised P groups results in the most active catalyst and epoxidises bulky olefins such as cyclooctene, norbornene and geraniol with good conversions and high selectivities (>93%).
Another interesting report19 of heterogeneous tungsten catalysts has demonstrated the immobilisation of peroxotungsten complexes on porous polymers modified with organophosphorous ligands. Peroxotungstic complexes linked to polystyrene grafted phosphoramides (1) (Fig. 3) catalyse the epoxidation of cyclohexene with high conversion (90%) and a high TOF of 400 (turnover frequency = mole of epoxide formed per mole of tungsten per h), but moderate selectivity (70%). Higher catalytic efficiency has been obtained for the polymethacrylate-grafted phosphotriamides (2) (Fig. 3) supported peroxotungstic complexes (TOF = 1000) in the epoxidation of cyclohexene. Those catalysts show not only higher activities but also better selectivities than the soluble complexes.
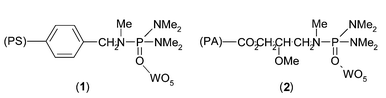 | ||
| Fig. 3 Polystyrene-grafted phosphoramides and polymethacrylate-grafted phosphotriamide. | ||
The new concept of triphase catalysis has been reported recently20 involving incorporating inorganic species in an amphiphilic chain polymer to form a self-assembled and networked supramolecular complex in water. A mixture of phosphotungstic acid and poly(N-isopropylacrylamide) copolymer containing quaternary ammonium salts has been used to prepare the triphasic catalyst where the quaternary ammonium salt moiety is possibly cross-linked by trivalent [PW12O40]−. The insoluble catalyst is very efficient in the epoxidation of allylic alcohols at room temperature and in the absence of solvent.
Polyoxometalates of the formula [ZnWMn2II(ZnW9O34)2]12− have been supported21 on a modified silica containing quaternary ammonium functionalities introduced using the sol–gel technique (Fig. 4). The catalytic activity is significantly increased when the silica surface is modified with polyethylene oxide (PEO) and polypropylene oxide (PPO). An intermediate 10% PEO, 10% PPO silica provides the highest activity due to the optimal contact of the organic substrate and the aqueous oxidant at the balanced hydrophobic/hydrophilic catalyst surface. Cycloctene epoxidation using 30% H2O2 has been obtained in higher yields compared to the homogeneous conditions under solvent free conditions.
![Schematic representation of the [ZnWMn2II(ZnW9O34)2]12− polyoxometalate immobilised on functionalised silica Q+–PE(P)O–SiO2.](/image/article/2003/GC/b208925b/b208925b-f4.gif) | ||
| Fig. 4 Schematic representation of the [ZnWMn2II(ZnW9O34)2]12− polyoxometalate immobilised on functionalised silica Q+–PE(P)O–SiO2. | ||
Hydrophobic mesoporous silica gel chemically modified with Ph3SiOEt and Me2NCH(OCH2Ph)2 in a 1∶1 ratio has been recently reported22 as an effective support for tris(cetylpyridinium)(12-tungstophospate) [π-C5H5N(CH2)15CH3]3(PW12O40)] polyoxometalate catalyst. A variety of terminal, cyclic, highly substituted and aromatic olefins have been selectively epoxidised with high yields using this catalyst (>97%).
New heterogeneous catalysts, containing tungstate WO42− ions exchanged onto Ni,Al-layered double hydroxides, have been applied to the bromide-assisted epoxidation with H2O2.23 Initially, oxidative bromination of olefins takes place to form the bromohydrin intermediate followed by its dehydrobromination to give the epoxide product (Fig. 5) under mild reaction conditions (pH 6–7). Carrying out the reaction under monophasic conditions in a mixed CH3CN–H2O (3∶7) solvent system favours the epoxidation reaction. Even though turnover frequencies are higher than classical W-catalysed epoxidations, the catalyst efficiency is limited to the epoxidation of geminally di-, tri- and tetrasubstituted olefins.
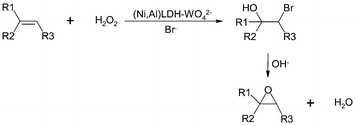 | ||
| Fig. 5 Bromide-assisted epoxidation of olefins with H2O2 catalysed by WO42−–Ni,Al-layered double hydroxides. | ||
Metalloporphyrins and metal salen complexes
The high activity of biological metalloporhyrins containing enzymes like cytochrome P450 has led to the development of porphyrin based catalysts for the enantioselective epoxidation of alkenes.24 The extensive research of metalloporphyrins catalysed epoxidation reactions has concentrated on using iron(III) and manganese(III) and the role of pyridine or imidazole ligands.25 The proposed mechanism for the reaction proceeds through the formation of a high valent metal–oxo species.26The use of hydrogen peroxide as an oxidant in metalloporphyrin catalysed epoxidations causes fast destruction of the catalysts, resulting in the dismutation of the oxidant. Furthermore, the structure of the catalyst has an important effect on its activity. A lot of work has been done to investigate the effect of the electronic and steric characteristics of the tetraphenylporphyrin on the stability and activity of those catalysts.27–30 In addition, the use of co-catalysts significantly affects the catalyst activity.
Addition of carboxylic acids to the manganese–porphyrin/hydrogen peroxide epoxidations leads to a remarkable increase in the reaction rates.31 A metal–acylperoxo complex, as shown in Fig. 6, was proposed to be a possible intermediate when acid was added in excess. Reaction rates strongly depend on both the amount of the acid and its acidity.
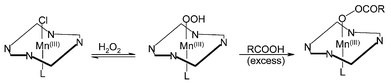 | ||
| Fig. 6 Proposed mechanism for the formation of a metallo–acylperoxo intermediate in the presence of an excess of the acid co-catalyst. | ||
Unsaturated monoterpenes have been efficiently epoxidised with hydrogen peroxide and ammonium acetate as a co-catalyst in the presence of MnIII porphyrin complexes.32
Enantioselective epoxidation of non-functionalised alkenes catalysed by a dimeric homochiral MnIII –Schiff base complex and urea–hydrogen peroxide as an oxidant has been recently reported33 in a DCM–MeOH solvent system and with ammonium acetate as a co-catalyst (Fig. 7). Complete conversions were obtained for a series of chromenes and the chiral induction increases for the electron deficient alkenes. The reaction rate decreases in the presence of other nitrogen or oxygen coordinating co-catalysts compared to ammonium acetate. Recycling of the catalyst reduces reaction rates while enantioselectivities remain constant.
 | ||
| Fig. 7 Manganese–salen catalysed epoxidation of olefins with urea–hydrogen peroxide as terminal oxidant and ammonium acetate as a co-catalyst. | ||
Catalytic epoxidation of olefins by an electron-rich FeIII porphyrin complex and hydrogen peroxide in aprotic solvents can be carried out effectively in the presence of 5-chloro-1-methylimidazole.34 The role of this electron poor imidazole is critical for the reaction as it decelerates the O–O bond cleavage of the reactive intermediate (Por)FeIII(OOR) complex.
The employment of micellar catalysis is advantageous for the manganese complexes catalysed epoxidation of terminal alkenes using hydrogen peroxide as the terminal oxidant.35 The homogeneous manganese porphyrin complex chloro(5,10,15,20-tetrakis(o-chlorophenyl)porphyrinato)manganese(III) solubilised in Triton X-100 (C(CH3)3CH2C(CH3)2C6H4(polyethylene oxide)9–10) in the presence of imidazole as a co-catalyst showed very good stability and selectivity in the epoxidation of 1-octene, however the conversion is quite low (25%).
The effect of different carboxylate salts on the epoxidation of 1,2-dihydronaphthalene with a manganese chiral salen complex and aqueous hydrogen peroxide was investigated.36 The highest reactivity was obtained for ammonium acetate. The exact role of the carboxylate in the system is not clear: promoting the formation of HO2−, acting as an axial ligand or forming peroxyacylmanganese species. It is evident however, that these salts significantly promote the reaction rates and selectivities of the reaction.
A significant increase of reaction reactivity and selectivity can be obtained by the addition of carboxylic acid anhydrides to a manganese salen complex–N-methylmorpholine N-oxide catalytic system.37 Peroxycarboxylic acids are formed in situ from anhydrous Ph3PO–H2O2 and carboxylic acid anhydrides while the highest enantioselectivity has been obtained with maleic anhydride as a co-catalyst.
Furthermore, even simple manganese salts such as MnSO4 have been reported38 to catalyse the epoxidation of a range of disubstituted alkenes with good yields in a hydrogen peroxide buffer solution (0.2 M, pH 8.0, NaHCO3). Percarbonate is formed in situ and combines with manganese to give the active intermediate, reducing the reaction times. However the method suffers from the use of DMF as a solvent and the large amount (10 equiv.) of hydrogen peroxide solution needed, which requires slow and careful addition.
Much effort has been made on immobilising manganese complexes on a solid support, as the homogeneous catalysts can not be recycled and they are rather expensive and unstable in contact with the oxidant. Catalysts covalently attached to silica have been prepared39 by grafting (5-(pentafluorophenyl)-10,15,20-tris(2,6-dichlorophenyl)porphyrinato)manganese(III) or (2,3,7,8,12,13,17,18-octachloro-5-(pentafluorophenyl)-10,15-20-tris(2.6-dichlorophenyl)porphyrinato)manganese(II) (Fig. 8) on aminopropylated silica. High yields for the epoxidation of cyclooctene with iodosylbenzene were obtained using those heterogeneous catalysts (95% and 67% respectively) and the first catalyst can be recycled giving a somewhat reduced but reproducible yield of 70%. Epoxidation of cyclohexene is less selective but it still gives the epoxide as the main product. Use of hydrogen peroxide as an oxidant shows very low yields in the epoxidation of cyclooctene, however, the reaction is enhanced by the addition of a weak base such as imidazole or ammonium acetate.
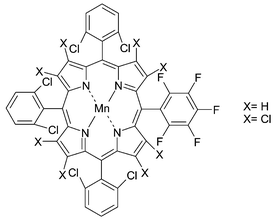 | ||
| Fig. 8 Manganese–porphyrin catalysts. | ||
Another active catalyst for the epoxidation of alkenes with hydrogen peroxide is the manganese(IV) complex based on the N,N′,N″-trimethyl-1,4,7-triazacyclononane (tmtacn) ligand. The use of this catalyst initially suffered from decomposition of hydrogen peroxide into water and O2.40 A significant improvement in the system came by the addition of catalytic amounts of ligands.41 A 1∶1 mixture of oxalic acid–oxalate buffer added to the system results in high conversions (95%) and selectivities of the epoxidation of terminal alkenes. Electron deficient alkenes react slightly faster than electron rich alkenes and the system can be used for the double epoxidation of (non)-conjugated dienes (Fig. 9).
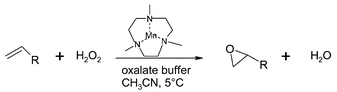 | ||
| Fig. 9 Mn–trimethyltriazacyclonane catalysed epoxidation of terminal olefins in the presence of an oxalate buffer. | ||
A very efficient manganese catalyst [Mn2O3(tmtacn)2](PF6)2 has been recently reported42 with hydroxymethoxyacetic acid methyl ester as a co-catalyst. High conversions have been obtained for the epoxidation of different cyclic olefins and turnover numbers to the epoxides were up to 600. However, substantial cis-diol products are formed depending on the ring size of alkene.
Novel dinucleating ligands incorporating the three N-donor atoms for each Mn atom have been synthesised43 and showed high turnovers of up to 900 for cyclohexene epoxidation. The catalyst is more reactive for disubstituted olefins than terminal alkenes. Even though reaction conditions are mild (ambient temperature), the employment of acetone as a solvent is hazardous due to the possible formation of acetone peroxide.
Non-heme iron complexes based on a tetradentate ligand (N,N′-dimethyl-N,N′-bis(pyridylmethyl)ethane-1,2-diamine), have been recently reported44 to epoxidise aliphatic alkenes at high yields (60–90%) with 50% hydrogen peroxide. The iron-complex mimics the oxidative enzyme methane monooxygenase. The addition of acetic acid (30 mol%) significantly favours the epoxide production due to the formation of a μ-oxo, carboxylate-bridged diiron(III) complex. This complex is easily formed in situ and produces a very active system, in which the epoxidation reaction occurs within 5 min.
Metal oxides
Methyltrioxorhenium (MTO) is an active catalyst for the epoxidation of alkenes using hydrogen peroxide as an oxidant. Initially, anhydrous hydrogen peroxide in ButOH was used as the oxidant but low selectivities were reported for acid sensitive epoxides due to the high acidity of the MTO–H2O2 system.45The addition of a base, such as pyridine,46 in large excess to MTO (>10∶1) enhances the rate and selectivity of the epoxidation of di-, tri- and tetra-substituted alkenes, as it protects the epoxide from ring opening. Commercially available aqueous 30% H2O2 can be used as an oxidant.
Epoxidation of unreactive terminal alkenes is achieved in the presence of the less basic 3-cyanopyridine or 3-fluoropyridine at high yields.47,48 Nevertheless, epoxidation of styrene requires a mixture of pyridine and 3-cyanopyridine to obtain both high conversion and protection of the styrene oxide. Use of pyrazole (24∶1 ratio) as the basic ligand and dichloromethane as solvent was reported to give even better results than the other bases.49
The system can be improved further by the use of fluorinated alcohols as solvents.50 The use of trifluoroethanol and MTO–pyrazole system with a low catalyst loading (1%), is very effective for the epoxidation a range of alkenes (yields >98%), with the exception of the very apolar ones (C12 or higher alkenes and stilbenes) which are insoluble in the reaction medium.
While hydrogen peroxide is typically used in the MTO catalysed epoxidation, other oxidants such as sodium percarbonate have been reported51 to act as an oxygen source. High yields were obtained for a range of alkene epoxidations catalysed by MTO–pyrazole and in the presence of trifluoroacetic acid. No external cooling is required as the interaction between the sodium percarbonate and the acid ensures the slow release of hydrogen peroxide.
MTO is a very active epoxidation catalyst, however, its difficult, hazardous and environmentally unfriendly synthesis involving organotin reagents52 makes it an unattractive option for epoxidation reactions.
Epoxidations in the absence of metals
The activation of hyrogen peroxide in the absence of metals by converting it in a more reactive oxidant has been the subject of many studies.Potassium peroxomonosulfate has mainly been used for the generation of dioxirane, as the sulfate moiety is a good leaving group, which facilitates the ring closure for the formation of dioxirane.53,54 However, hydrogen peroxide has been demonstrated55 to be an effective oxidant for the fructose derived dioxirane catalysed epoxidation of a number of olefins at high yields and ee’s. The use of acetonitrile is critical for the reaction, suggesting that the actual oxidant responsible for the in situ formation of dioxirane, is the corresponding peroxyimidic acid as shown in Fig. 10.
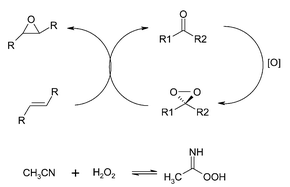 | ||
| Fig. 10 Epoxidation of alkenes by dioxiranes likely formed by peroxyimidic acid. | ||
The use of H2O2–CH3CN as an oxidative system has been explored56 in the presence of different achiral ketones. Among them, trifluoroacetone is the most active one (used in small amounts 10–30 mol%), for the epoxidation of a variety of terminal, cyclic, acyclic, trans, cis and trisubstituted olefins with good yields. The pH has a major effect on the reaction and the maximum epoxide yield was obtained at pH 10.
Another example of promoting the activity of hydrogen peroxide is by using carbodiimide with mildly basic or acidic catalysts. A peroxyisourea is possibly formed as a reactive intermediate by the reaction of hydrogen peroxide with an excess of of dicyclohexylcarbodiimide (DCC) (2 equiv.)57 as shown in Fig. 11. Hydroxyl containing solvents such as methanol or ethanol are preferable for the reaction. Disubstituted olefins are epoxidised in good yields, however reaction times are long and the selectivity of the reaction for substrates containing two double bonds is very poor. Furthermore after the reaction, equivalent amounts of urea compounds are formed which can not be easily recycled.
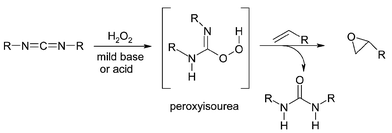 | ||
| Fig. 11 Activation of hydrogen peroxide in the presence of DCC by the formation of the intermediate peroxyisourea. | ||
An interesting method for activating hydrogen peroxide is through the presence of the hydrogen carbonate ion which forms the active oxidant peroxomonocarbonate ion, HCO4−.58 Water-soluble alkenes were epoxidised with stoichiometric amounts of hydrogen peroxide (1.5–6 equiv.) with good yields. Water insoluble alkenes can react under the same conditions if an acetonitrile–water (3∶2 v∶v) mixture is used as the solvent. The reaction conditions are mild, however conversions are low for less nucleophilic substrates and sensitive epoxides are readily hydrolysed under the reaction conditions.
Recently, a ‘green epoxidation system’ has been reported59 based on the use of CO2 as a solvent and hydrogen peroxide as the terminal oxidant in the presence of a base. Percarbonate is believed to be the active species and transfer limitation problems between the H2O/CO2 phases were overcome by the employment of surfactants. However, product yields are quite low, especially for n-alkenes.
Perfluorinated ketones combined with hydrogen peroxide have been demonstrated to enable epoxidation reactions. Perfluoroheptadecan-9-one is a very active catalyst,60 however a mixture of refluxing 1,2-dichloroethane–ethyl acetate is required to ensure its reasonable solubility in the system. Good product yields are obtained for a range of substrates and moderate yields are obtained for acid sensitive epoxides when the reaction mixture was buffered. Trifluoroethanol can be used as a solvent for the reaction but the recovery of the catalyst is poorer than in dichloroethane.
The use of fluorinated ketones suffers from environmental issues due to the high toxicity of these compounds. Immobilisation of fluorinated ketones is advantageous and a heterogeneous silicate synthesised by the sol–gel method containing hexafluoroketone has been efficiently used in the epoxidation of nucleophilic alkenes.61
Fluorinated alcohols such as trifluoroethanol and hexafluoro-2-propanol have been used as solvents62 for the uncatalysed epoxidation of a range of alkenes with an aqueous hydrogen peroxide buffered solution (Fig. 12). Trisubstituted alkenes were obtained in high yields (80–90%) by reflux in trifluoroethanol, except for hydrolysis-sensitive alkenes. Hexafluoro-2-propanol can be used at room temperature, giving the same rates as in boiling trifluoroethanol and in good yields for electron rich alkenes.
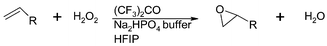 | ||
| Fig. 12 Epoxidation of olefins by the hexafluoroacetone–hydrogen peroxide system in hexafluoro-2-propanol. | ||
Heterogeneous catalysts
Heterogeneous catalysts offer the advantages of easy catalyst separation and sometimes higher selectivity, however most of these catalysts suffer from lower activity, instability in the epoxidation systems and limited recyclability.Titanium containing silicates, including amorphous titania–silica materials and Ti-substituted molecular sieves are the most efficient heterogeneous catalysts for epoxidation reactions.
A well-known solid epoxidation catalyst is the molecular sieve, type Ti Silicate-1 (TS-1)63 which can activate hydrogen peroxide as an oxidant and catalyse the epoxidation of different alkenes. The main drawback of this catalyst is its small pore dimensions of 5.6 × 5.3 Å, which makes it accessible to only relatively small reactants64 and restricts this catalyst to epoxidation of linear alkenes.
To overcome the steric limitation of TS-1, other mesoporous and macroporous, titanium containing zeolitic-materials have been developed, such as Ti,Al-beta, which has a three-dimensional pore structure. The presence of aluminium causes Brønsted acidity, which can lead to acid catalysed side reactions65 which thus decreases the selectivity of epoxide production.
Therefore, an aluminium-free Ti-beta catalyst has been developed66 which has been shown to efficiently catalyse the epoxidation of bulky substrates such as norbornene, substituted cyclohexenes, cyclic terpenes and allylic alcohols with hydrogen peroxide. For linear alkenes, the catalyst activity is lower than that of TS-1 and internal alkenes react faster than alkenes with a terminal double bond. For bulky substrates however, steric effects play an important role and contribute to the reactivity of the substrates, suggesting that the hindered formation of the transition state at the titanium site is an important factor in the activity of the catalyst.
Improvement of the activity and stability of Ti–silicate catalysts can be obtained by enhancing their hydrophobicity. Trimethylsilation of micro- and mesoporous titanosilicates by N,O-bis(trimethylsilyl)trifluoroacetamide increases the activity of the catalyst, particularly for the MCM and mixed SiO2/TiO2 aerogels. 67
Hydrophobic mesoporous titanosilicates can be prepared by the sol–gel technique using hexadecyltrimethylammonium chloride as a templating surfactant under acidic conditions during long gelation times. Temperatures lower than room temperature and a 0.014 H+/Si ratio proved to be beneficial for higher surface area and pore size of the material and a 2.5% Ti loading can be obtained.68 The catalysts enable the epoxidation of cyclohexene and norbornene at high selectivities (>95%) using TBHP or hydrogen peroxide as an oxidant.
Ti has been supported on amphiphilic NaY zeolite which have been partially modified with alkylsilyl groups.69 The surface of this catalyst contains both hydroxyl and alkylsilyl groups which results in their placement between hydrogen peroxide and the organic phase in the liquid–liquid phase boundary. Epoxidation of linear alkenes is efficiently catalysed and epoxide yields are higher under static conditions.
A unique trans-selectivity in the epoxidation of cis, trans alkenes has been shown70 for Ti-MWW zeolite (known as MCM-22) heterogeneous catalyst compared to the TS-1 and Ti-Beta. Selectivities reach up to 80% for the corresponding trans epoxide from a 50∶50 cis/trans ratio of the alkene. The increased trans selectivity is attributed to the structure of the Ti-MWW, specifically, the 10-MR (membered ring) sinusoidal channels whose shape promote the epoxidation of trans alkenes.
Rhenium oxides Re2O7 and ReO4− have been supported on zeolite Y, mixed alumina–silica and pure alumina as epoxidation catalysts with anhydrous hydrogen peroxide.71 Stable catalysts are formed in the case of the alumina support and ReO4−. The schematic representation of the activation of the immobilised MTO in alumina by hydrogen peroxide is shown in Fig. 13. Low conversions (40%) were obtained for the epoxidation of cyclooctene, while selectivity is high (>96%). Epoxidation of cyclohexene using these catalysts failed as cyclohexane diol is obtained as the major product.
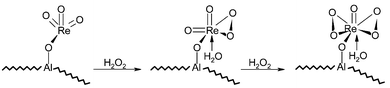 | ||
| Fig. 13 Reaction of hydrogen peroxide with the immobilised ReO3 to form the active complexes. | ||
Ordinary chromatographic alumina has been shown to have good catalytic activity in the epoxidation of a range of 1-alkylcyclohexenes using hydrogen peroxide which is dried in situ by performing the reaction under reflux with Dean–Stark water separation.72 Completely anhydrous conditions result in lower selectivity, as the alumina catalysed decomposition of the epoxide and hydrogen peroxide is prevented by the presence of only a small amount of water. The system is not very reactive however, for the epoxidation of cycloalkenes.73
Hydrotalcites Mg10Al2(OH)24(CO3) have been reported74 as efficient heterogeneous base catalysts for the epoxidation of various olefins using hydrogen peroxide as an oxidant and isobutyramide as a co-catalyst. Addition of an anionic surfactant, sodium dodecyl sulfate, remarkably enhances the rate of the reaction, while cationic surfactants inhibit the reaction and non-ionic surfactants have no effect. Hydrocarbons can be used as solvents, even though 1,2-dichloroethane gives higher conversions for cyclooctene epoxidation. The catalyst can be reused without a significant loss of catalytic activity. However, the method suffers due to the use of equivalent amounts of amide, which are consumed during the reaction to the corresponding acids.
Electron deficient α,β-unsaturated ketones have been efficiently (>90%) epoxidised under mild conditions by hydrogen peroxide in the presence of basic hydrotalcite Mg10Al2(OH)24(CO3).75 Methanol can be used as a solvent and excellent hydrogen peroxide utilisation can be attained. The presence of quaternary ammonium salts as phase transfer catalysts enhance the reaction, especially for hydrophobic ketones. The catalyst actually promotes the transformation of hydrogen peroxide into the perhydroxyl anion as the active species and can easily be recycled and reused without the loss of their activity. Heating by microwave radiation in the hydrotalcite catalysed epoxidation of olefins with hydrogen peroxide and a nitrile, accelerates significantly the reaction rates and results in higher yields of epoxides.76
Furthermore, the intercalation of anionic surfactants, such as dodecyl sulfate and dodecyl benzenesulfonate, into double layered hydroxide hydrotalcite results in a more selective catalyst for the epoxidation of limonene to its 1,2-epoxide. However, conversions are lower in comparison with the carbonate analogue and an excess of nitrile is necessary in order to activate the hydrogen peroxide.77
Conclusions
The development of new epoxidation methods based on the use of hydrogen peroxide as an oxidant in order to achieve the cleaner synthesis of epoxides has been described in this review. The use of metal catalysts is very important, however further development in order to replace the often-used chlorinated solvents and to more efficiently separate the catalysts is needed (possibly, through the use of nanofiltration membranes). Activation of hydrogen peroxide in the absence of metals results in the production of large amounts of waste due to the need for auxiliaries.The use of heterogenised homogeneous catalysts has had limited success due to poor product yields and catalyst stability, which leads to leaching during reactions and thus limits the potential for recyclability. While the use of hydrogen peroxide as an oxidant in epoxide reactions is an attractive method in the context of green chemistry, the employment of redox zeolites catalysts such as titanium–silicates (TS-1) is limited to linear alkenes.
Improving the stability of supported metal catalysts is generally an important goal in oxidation catalysis and the difficulty is increased by the necessity of an aqueous oxidant. Further research in the synthesis of new catalytic materials by maximising their accessible surface area and tuning their surface polarity and organophilicity is required to achieve more active epoxidation catalysts.
References
- K. Bauer, D. Garbe and H. Surburg, Common Fragrance and Flavor Materials, Wiley-VCH, New York/Weinheim, 1997 Search PubMed.
- G. Sienel, R. Rieth and K. T. Rowbottom, Ullmann’s Encyclopedia of Organic Chemicals, Wiley-VCH, Weinheim, 1999 Search PubMed.
- R. A. Van Santen and H. P. C. E. Kuipers, Adv. Catal., 1987, 35, 265 Search PubMed.
- R. A. Sheldon and J. K. Kochi, Metal-catalysed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- R. Landau, G. A. Sullivan and D. Brown, CHEMTECH, 1979, 602 Search PubMed.
- R. A. Sheldon, J. Mol. Catal., 1980, 7, 107 CrossRef.
- D. Swern, Organic Peroxides, ed. D. Swern, Wiley Interscience, New York, 1971, vol. 2 Search PubMed.
- C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407 CrossRef CAS.
- R. Neumann and M. Gara, J. Am. Chem. Soc., 1995, 117, 5066 CrossRef CAS.
- D. E. Katsoulis and M. T. Pope, J. Am. Chem. Soc., 1984, 106, 2737 CrossRef CAS.
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto and R. Noyori, J. Org. Chem., 1996, 61, 8310 CrossRef CAS.
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto, D. Panyella and R. Noyori, Bull. Chem. Soc. Jpn., 1997, 70, 905 CAS.
- X. Zuwei, Z. Ning, S. Yu and L. Kunlan, Science, 2001, 292, 1139 CrossRef CAS.
- R. Neumann, A. M. Khenkin, D. Juwiler, H. Miller and M. Gara, J. Mol. Catal., 1997, 117, 169 Search PubMed.
- M. Bösing, A. Nöh, I. Loose and B. Krebs, J. Am. Chem. Soc., 1998, 120, 7252 CrossRef.
- P. A. L. de Villa, B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Org. Chem., 1999, 64, 7267 CrossRef CAS.
- C. Venturello and R. D’Aloisio, J. Org. Chem., 1988, 53, 1553 CrossRef CAS.
- D. H. Hoegaerts, B. F. Sels, D. E. De Vos, F. Verpoort and P. A. Jacobs, Catal. Today, 2000, 60, 209 CrossRef CAS.
- G. Gebard, C. R. Acad. Sci. Paris, Série IIc, Chim./Chem., 2000, 3, 757 Search PubMed.
- Y. M. A. Yamada, M. Ichinohe, H. Takahashi and S. Ikegami, Org. Lett., 2001, 3, 1839 CrossRef CAS.
- M. Cohen and R. J. Neumann, J. Mol. Catal., 1999, 291 Search PubMed.
- T. Sakamoto and C. Pac, Tetrahedron Lett., 2000, 41, 10009 CrossRef CAS.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Am. Chem. Soc., 2001, 123, 8350 CrossRef CAS.
- R. A. Sheldon, Metalloporphyrins in Catalytic Oxidations, Marcel Dekker, New York, 1994 Search PubMed.
- J. P. Collman, X. Zhang, V. J. Lee, E. Uffelman and J. I. Brauman, Science, 1993, 261, 1404 CAS.
- L. A. Campbell and T. Kodadek, J. Mol. Catal., 1996, 113, 293 Search PubMed.
- S. Banfi, S. Montanari and S. Quici, J. Org. Chem., 1989, 54, 1850 CrossRef CAS.
- D. Mansuy, Coord. Chem. Rev., 1993, 125, 129 CrossRef CAS.
- S. Banfi, S. Montanari, S. Quici, S. V. Barkanova, O. L. Kaliya, U. N. Kopranenkov and E. A. Luk′Yanets, Tetrahedron Lett., 1995, 36, 2317 CrossRef CAS.
- M. D. Assis, A. J. B. Melo, O. A. Serra and Y. Iamamoto, J. Mol. Catal., 1995, 97, 41 Search PubMed.
- A. M. d’A. Rocha Gonsalves and A. C. Serra, J. Mol. Catal., 2001, 168, 25 Search PubMed.
- R. R. L. Martins, M. G. P. M. S. Neves, A. J. D. Silvestre, M. M. Q. Simões, A. M. S. Silva, A. C. Tomé, J. A. S. Cavaleiro, P. Tagliatesta and C. Crestini, J. Mol. Catal., 2001, 172, 33 Search PubMed.
- R. I. Kureshy, N. H. Khan, S. H. R. Abdi, S. T. Patel and R. V. Jasra, Tetrahedron: Asymmetry, 2001, 12, 433 CrossRef CAS.
- W. Nam, H. J. Lee, S. Y. Oh, C. Kim and H. G. Jang, J. Inorg. Biochem., 2000, 80, 219 CrossRef CAS.
- L. J. P. Van den Broeke, V. G. De Bruijn, J. H. M. Heijnen and J. T. F. Keurentjes, Ind. Eng. Chem. Res., 2001, 40, 5240 CrossRef CAS.
- P. Pietikäinen, Tetrahedron, 1998, 54, 4319 CrossRef CAS.
- P. Pietikäinen, J. Mol. Catal., 2001, 165, 73 Search PubMed.
- B. S. Lane and K. Burgess, J. Am. Chem. Soc., 2001, 2933 CrossRef CAS.
- F. G. Doro, J. R. Lindsay Smith, A. G. Ferreira and M. D. Assis, J. Mol. Catal., 2000, 164, 97 Search PubMed.
- R. Hage, J. E. Iburg, J. Kerschner, J. H. Koek, E. L. M. Lempers, R. J. Martens, U. S. Racherla, S. W. Russell, T. Swarthoff, M. R. P. van Vliet, J. B. Warnaar, L. van der Wolf and B. Krijnen, Nature, 1994, 369, 637 CrossRef CAS.
- D. E. De Vos, B. F. Sels, M. Reynaers, Y. V. Subba Rao and P. A. Jacobs, Tetrahedron Lett., 1998, 39, 3221 CrossRef CAS.
- J. Brinksma, L. Schmieder, G. van Vliet, R. Boaron, R. Hage, D. E. De Vos, P. L. Alsters and B. L. Feringa, Tetrahedron Lett., 2002, 43, 2619 CrossRef CAS.
- J. Brinksma, R. Hage, J. Kerschner and B. L. Feringa, Chem. Commun., 2000, 537 RSC.
- M. C. White, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 7194 CrossRef CAS.
- W. A. Herrmann, R. W. Fischer, M. U. Rauch and W. Scherer, J. Mol. Catal., 1994, 86, 243 CrossRef CAS.
- J. Rudolph, K. L. Reddy, J. P. Chiang and K. B. Sharpless, J. Am. Chem. Soc., 1997, 119, 6189 CrossRef CAS.
- C. Copéret, H. Adolfsson and K. B. Sharpless, Chem. Commun., 1997, 1565 RSC.
- H. Adolfsson, C. Copéret, J. P. Chiang and A. K. Yudin, J. Org. Chem., 2000, 65, 8651 CrossRef CAS.
- W. A. Herrmann, R. M. Kratzer, H. Ding, W. R. Thiel and H. Glas, J. Organomet. Chem., 1998, 555, 293 CrossRef CAS.
- M. C. A. Van Vliet, I. W. C. E. Arends and R. A. Sheldon, Chem. Commun., 1999, 821 RSC.
- A. R. Vaino, J. Org. Chem., 2000, 65, 4210 CrossRef CAS.
- W. A. Herrmann, R. M. Kratzer and R. W. Fischer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2652 CrossRef CAS.
- D. Yang, M. K. Wong and Y. C. Yip, J. Org. Chem., 1995, 60, 3887 CrossRef CAS.
- S. E. Denmark, D. C. Forbes, D. S. Hays, J. S. DePue and R. G. Wilde, J. Org. Chem., 1995, 60, 1391 CrossRef CAS.
- L. Shu and Y. Shi, Tetrahedron Lett., 1999, 40, 8721 CrossRef CAS.
- L. Shu and Y. Shi, J. Org. Chem., 2000, 65, 8807 CrossRef CAS.
- G. Majetich, R. Hicks, G. Sun and P. McGill, J. Org. Chem., 1998, 63, 2564 CrossRef CAS.
- H. Yao and D. E. Richardson, J. Am. Chem. Soc., 2000, 122, 3220 CrossRef CAS.
- D. Hâncu, J. Green and E. J. Beckman, Acc. Chem. Res., 2002, 35 CAS , 757.
- M. C. A. Van Vliet, I. W. C. E. Arends and R. A. Sheldon, Chem. Commun., 1999, 263 RSC.
- K. Neimann and R. Neumann, Chem. Commun., 2001, 487 RSC.
- M. C. A. Van Vliet, I. W. C. E. Arends and R. A. Sheldon, Synlett., 2001, 2, 248.
- M. Taramasso, G. Perego and B. Notari, US Pat., 4410501, 1983.
- M. G. Clerisi and P. Ingallina, J. Catal., 1993, 140, 71 CrossRef CAS.
- A. Corma, M. A. Camblor, P. Esteve, A. Martinez and J. Perez-Pariente, J. Catal., 1994, 145, 151 CrossRef CAS.
- J. H. Van der Waal, M. S. Rigutto and H. van Bekkum, Appl. Catal. A, 1998, 167, 331 CrossRef CAS.
- M. D’Amore and S. Schwarz, Chem. Commun., 1999, 121 RSC.
- F. Figueras, H. Kochkar and S. Caldarelli, Microporous Mesoporous Mater., 2000, 39, 249 CrossRef CAS.
- S. Ikeda, H. Nur, T. Sawadaishi, K. Ijiro, M. Shimomura and B. Ohtani, Langmuir, 2001, 17, 7976 CrossRef CAS.
- P. Wu and T. Tatsumi, J. Phys. Chem. B, 2002, 106, 748 CrossRef CAS.
- D. Mandelli, M. C. A. van Vliet, U. Arnold, R. A. Sheldon and U. Schuchardt, J. Mol. Catal., 2001, 168, 165 Search PubMed.
- M. C. A. Van Vliet, D. Mandelli, I. W. C. E. Arends, Ulf Schuchardt and R. Sheldon, Green Chem., 2001, 3, 243 RSC.
- D. Mandelli, M. C. A. van Vliet, R. Sheldon and Ulf Schuchardt, Appl. Catal. A: General, 2001, 219, 209 CrossRef CAS.
- K. Yamagushi, K. Ebitani and K. Kaneda, J. Org. Chem., 1999, 64, 2966 CrossRef CAS.
- K. Yamagushi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Org. Chem., 2000, 65, 6897 CrossRef CAS.
- U. R. Pillai, E. Sahle-Demessie and R. S. Varma, Tetrahedron Lett., 2002, 43, 2909 CrossRef CAS.
- M. A. Aramendia, V. Borau, C. Jiménez, J. M. Luque, J. M. Marinas, J. R. Ruiz and F. J. Urbano, Appl. Catal. A: General, 2001, 216, 257 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |