On-board hydrogen production in a hybrid electric vehicle by bio-ethanol oxidative steam reforming over Ni and noble metal based catalysts
V. Fierro†, O. Akdim and C. Mirodatos*
Institut de Recherches sur la Catalyse, CNRS, 69626, Villeurbanne, France. E-mail: mirodata@catalyse.univ-lyon1.fr
First published on 16th December 2002
Abstract
In this work we present the optimisation of the oxidative steam reforming of ethanol for hydrogen production in order to feed a Solid Polymer Fuel Cell (SPFC) over a Ni–Cu/SiO2 catalyst at on-board conditions. The optimised experimental conditions involve a reforming temperature close to 700 °C, a molar ratio of H2O/EtOH equal to 1.6 and a molar ratio of O2/EtOH equal to 0.68. These conditions were also used to test noble metal (Pt, Pd, Ru and Rh) based catalysts. Ni and Rh based catalysts were tested in ageing experiments for up to 140 h at the optimised operating conditions. Ni–Cu/SiO2 and Rh/Al2O3 catalysts allowed a high hydrogen mixture as well as a constant selectivity to the reaction products, moreover the ethanol was totally converted during this time. In contrast, the Ni/SiO2 catalyst showed a continuous decrease in hydrogen production during ageing. An explanation to the differences between Ni and Ni–Cu catalysts is given in terms of the lower coke deposition on Ni–Cu catalyst due to the alloy formed. The rich hydrogen content in the outlet flow from the reformer obtained with the Ni–Cu, Ru and Rh based catalysts can be considered of high interest for fuel cells (FC) in mobile applications.
Green ContextThe development of fuel cells for automotive use relies on the on-board production of hydrogen in the vehicle. Most effort has been put into the development of methods for the extraction of hydrogen from methanol, which has a H∶C ratio of 4, but less work has been published on the use of the less hydrogen rich ethanol. This paper shows that ethanol can be produced efficiently under conditions where no NOx or SOx emissions take place, and thus (bio) ethanol could be an appropriate fuel for fuel cell applications.DJM |
1 Introduction
Fuel cells (FCs) forego the traditional extraction of energy in the form of combustion heat, conversion of heat energy to mechanical energy and finally turning mechanical energy into electricity. FCs operate like continuous batteries when supplied with fuel (hydrogen) to the anode and oxidant (oxygen or air) to the cathode. Encouraged by a catalyst, the hydrogen atom splits into a proton and an electron, which take different paths to the cathode. Protons pass through the electrolyte and electrons create a separate current that can be utilised before they return to the cathode, to be reunited with the hydrogen and oxygen in a molecule of water. Therefore, FCs chemically combine the molecules of a fuel and oxidiser without burning but converting them directly to electrical energy and so dispensing with the inefficiencies and pollution of traditional combustion.According to the U.S. Department of Energy, ‘Solid Polymer Fuel Cells (SPFC) are the primary candidates for light-duty vehicles, for buildings, and potentially for much smaller applications such as replacements for rechargeable batteries’. These cells have high power density, can vary their output quickly to meet shifts in power demand, and so are suited for automobiles where quick startup is required.
Ideally, vehicles would store hydrogen fuel onboard in high-pressure tanks. However, because current technology does not permit storage of enough H2 to deliver the driving range to which motorists are accustomed, hydrogen may initially be supplied from liquid fuels. Fuels containing hydrogen generally require a ‘fuel reformer’ that extracts the hydrogen. Generally, the reformate gas include H2, CO, CO2, H2O and a small amount of CH4. Carbon monoxide needs to be completely converted both because it is a criterion pollutant and also because it poisons the platinum electrodes, limited to 100 ppm.1 Therefore other processes are required:2–4 water gas shift (WGS) reactions, and/or selective oxidation (SELOX). After energy production in SPFC, the residual amounts of hydrogen and byproducts are converted into carbon dioxide and water in the catalytic burner.
Hydrogen sources include fossil fuels as methanol, ethanol, natural gas, petroleum distillates, liquid propane and gasified coal and even gas from landfills and wastewater treatment plants. Alcohols exhibit high qualities as H2 generators, since they are easily decomposed in the presence of water (steam reforming reaction) and generate a H2-rich mixture suitable for feeding fuel cells. Ethanol appears as an attractive alternative to methanol since it is much less toxic and is already used as versatile transportation fuel that offers a high octane number, a high heat of vaporisation and a low photochemical reactivity. Moreover, bio-ethanol can be produced in large quantities from biomass fermentation, therefore as a renewable energy source. This alcohol has also a significant advantage over fossil-fuel based systems: it is CO2 neutral, since the carbon dioxide that is produced in the process is consumed by biomass growth and a closed carbon cycle is operated.
Several works have been recently published on ethanol steam reforming over Ni, Co, Ni/Cu and noble metals (Pd, Pt, Rh) on various supports.5–14 In contrast, little work has been done on oxidative steam reforming despite the fact that it has been shown as a very convenient method for the selective production of hydrogen from methane15 and methanol16 and that the process can be made more energy-efficient and the hardware can be smaller and lighter.17 In a previous work we presented the optimisation of oxidative steam reforming of ethanol at diluted conditions over a Ni–Cu/SiO2 catalyst.18 The objective of the present study is to investigate in detail the effect of operating variables of the oxidative steam reforming of ethanol over the same catalyst under on-board conditions. Moreover, the hydrogen yield and the reaction selectivities obtained with this catalyst are compared with those obtained with noble metal (Pt, Pd, Ru and Rh) based catalysts.
2 Experimental
2.1 Catalyst
The active and selective materials selected for ethanol steam reforming were two Ni based catalysts supported on SiO2, one of them doped with Cu, and several 5% noble metal (Pt, Pd, Ru, Rh) based catalysts supported on Al2O3 and supplied by Engelhardt. The Ni/SO2 and Ni–Cu/SiO2 catalysts were prepared on a silica support (Degussa) with a BET surface of 200 m2 g−1 and by ionic exchange using Ni(NO3)2·6H2O and Cu(NO3)2·3H2O as precursors. The Ni/SiO2 catalyst has a Ni content of 19.4%. The Ni–Cu/SiO2catalyst has a Cu/(Ni + Cu) weight ratio of 10 and a total metal content of 18.4%. After calcination at 650 °C for 15 h, the Ni based catalysts were sieved to 0.2–0.3 mm.2.2 Catalytic tests
Prior to catalytic testing, the catalyst was placed in a fixed bed reactor and reduced under flowing hydrogen (30 ml min−1) at 650 °C for 8 h with a heating rate of 2 °C min−1. After reduction the catalyst was cooled down to reaction temperature under helium atmosphere. The runs were performed under on-board conditions with helium instead of nitrogen at atmospheric pressure in a fixed bed reactor (ID = 4 mm and hbed = 7 mm) where 50 mg of the catalyst are introduced. Catalyst was dispersed with SiC to minimise hot spot effects.Two parallel reactors and a system of valves allow pretreating a catalyst while a second one is being tested. There are two lines of gases to the reactors providing an ethanol–water–air-like (79% He and 21% O2) mixture from a gaseous He–O2 mixture and a liquid H2O–EtOH blend or a gas flow to pre-treat the catalyst with H2. The H2O–EtOH blend is regulated by a HPLC pump, the liquid is vaporised at 130 °C and then mixed with the gas flow before being fed to the fixed bed reactor or being sent to analysis before reaction. Gases were analysed on line by mass spectrometry and by gas chromatography. Wet gases were analysed by means of a Hewlett-Packard gas chromatograph model 6890, equipped with a TCD detector and a HP-PlotQ column that analyses CO2, CO, CH4 and other hydrocarbons as C2H6, C2H4 as well as EtOH and H2O. Helium was used as the internal standard and the variation of its concentration measured by gas spectrometry allows the evaluation of the volume correction factor and of the ethanol conversion and selectivities to the reaction products.
2.3 Kinetic parameter formulae
Since this work was carried out at on-board conditions a volume correction factor (CF) that takes into account the volume change as a result of reactions was considered to calculate conversions and selectivities.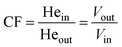 | (1) |
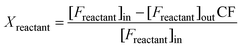 | (2) |
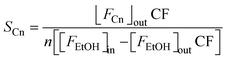 | (3) |
Cdeposited = ([Fcarbon inVin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −Fcarbon outVout] × 12)/mcatalyst −Fcarbon outVout] × 12)/mcatalyst | (4) |
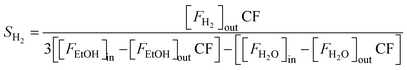 | (5) |
| tc = mcatal (kg)/EtOH (mol min−1) | (6) |
2.4 Ageing experiments
Ageing experiments were carried out using 50 mg of catalyst, a H2O–EtOH molar ratio of 1.6 and O2/EtOH molar ratio of 0.68 at 700 °C and a total volume flow of 80 ml min−1. Three catalysts were tested under these conditions, the two Ni catalysts prepared in our laboratory and the 5% Rh/Al2O3 commercial catalyst.3 Results and discussion
The oxidative steam reforming of ethanol is an intermediate step of a global process that allows electricity production. The optimisation criteria of the operating parameters must take into account the technologic limitations fixed by the system: (1) limitation of H2O/EtOH molar to minimise the volume and weight of the system; (2) reduction of carbon deposition on the catalyst to prolong the lifetime of the catalyst and (3) reduction of the CO concentration to limit the importance (weight and volume) of the WGS and SELOX steps downstream.Homogeneous reactions are very important when studying the oxidative steam reforming of ethanol and special attention must be paid to reduce the dead volume to avoid them. Fig. 1 shows the selectivity of the ethanol reforming in absence of catalyst at temperatures from 450 to 800 °C, with a H2O/EtOH molar ratio of 1.6 and a O2/EtOH molar ratio of 0.68. Ethanol decomposes at temperatures higher than 450 °C reaching a conversion of 95% at 700 °C with a total oxygen conversion from 550 °C. The selectivity to the reaction products is nearly constant at temperatures higher than 500 °C. There is a high selectivity to CO (≈50%) and the selectivity to hydrogen remains very low (≈30%) due to the high reaction selectivities to hydrogenated products as methane, ethane and ethylene and the production of water by the EtOH combustion. The selectivity to C2H4 (≈18%) is also rather important, ethylene acts as a very strong promoter of carbon formation and probably an important quantity of ethanol is converted to coke.
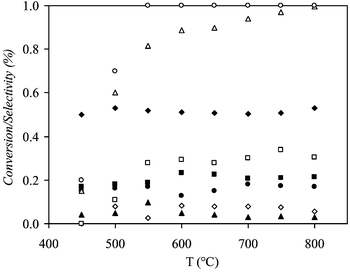 | ||
| Fig. 1 Conversion of ethanol and oxygen and product selectivity of ethanol reforming reaction versus temperature at homogenous conditions; O2/EtOH = 0.68, H2O/EtOH = 1.6 and flow rate = 80 cm3 min−1: (◆) SCO, (■) SCH4, (▲) SCO2, (●) SC2H4, (◇) SC2H6, (□) SH2, (△) XEtOH, (○) XO2. | ||
In a previous work we optimised the operating variables for ethanol oxidative reforming at diluted conditions. A temperature close to 600 °C, a H2O/EtOH molar ratio 1.55 and a O2/EtOH molar ratio of 0.5 with a contact time close to 1 min kg mol−1 resulted in the highest selectivity to hydrogen (92.5%) reducing in turn the amount of water introduced and the carbon deposition.
Fig. 2 shows the effect of temperature on reactants conversion and products selectivity at on-board conditions using the H2O/EtOH and O2/EtOH molar ratios optimised at diluted conditions, 1.55 and 0.5, respectively. Ethanol is completely converted over the whole studied temperature range and the conversion of water increases with temperature. The selectivity toward H2 and CO increases with temperature, whereas the selectivity toward CH4 decreases. Selectivity to hydrogen is only of 61.6%, this change (compared with 92.5% at diluted conditions) probably indicates a complex kinetic law with positive and negative partial reaction orders that are changed due to the higher partial pressures of the reactants at on-board conditions. Therefore, reaction temperature has to be increased in order to recover an important hydrogen production at on-board conditions. A reaction temperature close to 700 °C allows having an important production of hydrogen keeping the CO2/COx ratio high enough for the downstream processes of the on-board system.
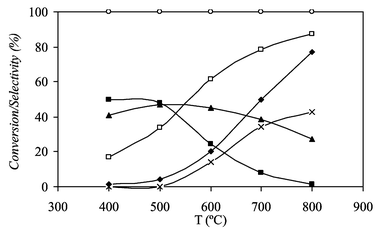 | ||
| Fig. 2 Effect of temperature on reactants conversion and products selectivity at on-board conditions; H2O/EtOH = 1.55 and O2/EtOH = 0.5: (◆) SCO, (■) SCH4, (▲) SCO2, (□) SH2, (×) XH2O, (○) XO2. | ||
Fig. 3 shows the evolution of reactant conversions and reaction selectivities as the O2/EtOH molar ratio increases at 700 °C. The introduction of a larger amount of oxygen reduces selectivities to CH4 and CO whereas CO2 and H2O are produced in a larger extent. It appears as if increasing the O2/EtOH ratio, O2 is used to oxidise CH4 and part of the CO. Selectivity to hydrogen is also improved (96.8%) by increasing air feeding to the system. Moreover the introduction of a higher quantity of oxygen reduces the carbon deposition and so increases the lifetime of the catalyst. Under these more realistic conditions the optimised experimental conditions involve a reforming temperature close to 700 °C, a molar ratio of H2O/EtOH equal to 1.6, a molar ratio of O2/EtOH equal to 0.68 and a contact time close to 0.2 min kg mol−1.19 The composition of the outlet flow from the reformer was H2/CO/CO2/CH4/H2O/N2 = 33/13/9/1/13/31.
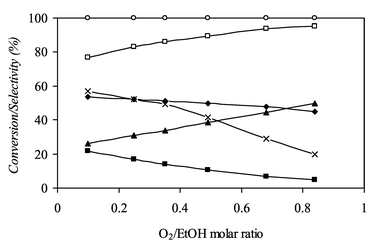 | ||
| Fig. 3 Evolution of reactant conversions and reaction selectivities versus O2/EtOH molar ratio at 700 °C: (◆) SCO, (■) SCH4, (▲) SCO2, (□) SH2, (×) XH2O, (○) XO2. | ||
3.1 Noble metal catalysts
5% Pd, Pt, Ru and Rh on Al2O3 were tested at temperatures from 650 to 800 °C, a molar ratio of H2O/EtOH equal to 1.6, a molar ratio of O2/EtOH equal to 0.68 and a total volume flow of 80 cm3 min−1. We have already discussed the mechanism of the ethanol reforming as a function of the temperature domain with a Ni–Cu catalyst.18 We showed that the acetaldehyde formation results from ethanol dehydrogenation at 300 °C and in the absence of oxygen. However, when catalysts based on Pd and Pt were tested there was a very high selectivity to acetaldehyde and high yields to CH4 even at high temperatures. The amounts of C2H6 and C2H4 were also important and so the selectivity of ethanol reforming to hydrogen was rather small, never higher than 50–60%. Fig. 4(a) shows the product selectivities and the ethanol conversion for the oxidative ethanol reforming when using a 5% Pt/Al2O3 catalyst. The oxidative-dehydrogenation of ethanol on Pd and Pt based catalysts has an important role up to 750 °C. Selectivity to acetaldehyde (Sacet) decreases as reaction temperature increases being 50 and 20% at 650 and 700 °C, respectively. At 750 °C the decomposition of acetaldehyde is complete and CH4 reforming and the water gas shift (WGS) reactions control the selectivities of the reaction.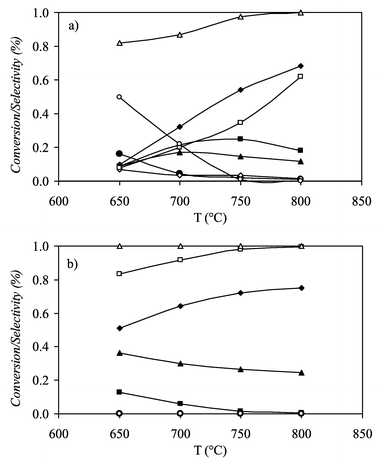 | ||
| Fig. 4 Product selectivity reforming reaction and conversion of ethanol versus temperature (a) for 5% Pt/Al2O3 catalyst and (b) for 5% Rh/Al2O3 catalyst; O2/EtOH = 0.68, H2O/EtOH = 1.6 and tc= 0.084 min kg mol−1: (◆) SCO, (■) SCH4, (▲) SCO2, (●) SC2H4, (◇) SC2H6, (□) SH2, (△) XEtOH, (○) Sacet. | ||
Ru and Rh based catalysts showed much better performances. Fig. 4(b) shows the product selectivities and the ethanol conversion for the oxidative ethanol reforming when using a 5% Rh/Al2O3 catalyst. Selectivities to hydrogen higher than 70% were already reached at 650 °C. Performances improved with reaction temperature and selectivity to CH4 was negligible at 750 °C. No ethylene production was observed in the temperature range of study and so a lower deposition of coke can be expected.
An order in the performance of the noble metal based catalysts can be established based on the experiments carried out at the standard conditions: Pt < Pd ≪ Ru < Rh. Fig. 5 shows the results observed with the 5% noble metal/Al2O3 at 700 °C. On the one hand Pt and Pd show similar performances and low selectivities to H2 (0.20 and 0.23%, respectively), whilst Ru and Rh showed much higher selectivities to H2 (0.75 and 0.90%, respectively). The very different results observed from Pd and Pt to Ru and Rh could be explained by the existence of a different reaction mechanism. In this sense, a detailed mechanism study by IR-DRIFT is in progress in our group.
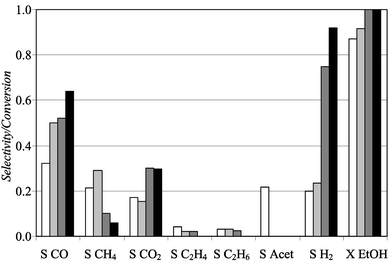 | ||
Fig. 5 Product selectivities of the reforming reaction and conversion of ethanol at 700 °C for noble metal based catalysts on Al2O3; O2/EtOH = 0.68, H2O/EtOH = 1.6 and tc = 0.084 min kg mol−1: (□) 5% Pt, (![[square tinted (light)]](https://www.rsc.org/images/entities/char_e068.gif) ) 5% Pd, ( ) 5% Pd, (![[square tinted (heavy)]](https://www.rsc.org/images/entities/char_e069.gif) ); 5% Ru, (■) 5% Rh. ); 5% Ru, (■) 5% Rh. | ||
3.2 Ageing experiments
Since the commercial development of oxidative reforming of ethanol requires a good stability of the catalyst, the most promising catalysts Ni–Cu/SiO2 and 5% Rh/Al2O3 catalysts were tested for 140 h at the optimised operating conditions. Their selectivity toward hydrogen initially decreased with time-on-stream and stabilised after 10 h at values at 89 and 88% for Ni–Cu/SiO2 and 5% Rh/Al2O3, respectively. These catalysts exhibited also a constant selectivity to the rest of reaction products and the ethanol was totally converted during this time.Most workers agree on a metastable metal carbide being an intermediate stage in the growth of filamentary carbon. Formation of this metal carbide can be avoided by alloying the active metal with a metal not forming a carbide of significant stability. The doping of copper adjusts the properties of nickel particles, alters their existing state and also changes their affinity with carbon. The melting points of Ni and Cu are 1455 and 1084 °C, respectively, and that of Ni–Cu alloy is between that of Ni and Cu. Rostrup-Nielsen et al.20 stated that small amounts of Cu alloying promotes while larger amounts (Cu∶Ni = 0.1) inhibits carbon formation and changes the morphology of the filaments.
A Ni/SiO2 catalyst was also tested and results compared with the Ni–Cu/SiO2 catalyst in order to study the efficiency of Cu addition. This catalyst deactivated progressively with time-on-stream and the ageing experiment was stopped after 100 h when selectivity towards hydrogen was 65%. As selectivity towards H2 decreased C2 species such as acetaldehyde and ethylene increased and deposition of coke became significant due either to direct ethanol dissociation or to the Boudard reaction. Although catalyst performance is initially reduced by Cu addition, we have shown that it considerably increases the lifetime of the catalyst for the oxidative steam reforming of ethanol at on-board conditions. Furthermore, copper allows total conversion of ethanol and constant selectivity towards hydrogen.
4 Conclusions
Results reported here clearly indicate that high efficiency and nearly zero emissions characterise oxidative steam reforming of bioethanol. Ni–Cu/SiO2 and Rh/Al2O3 catalysts show high activities and selectivities toward hydrogen production in contrast to Ni/SiO2 catalyst that deactivates rapidly due to coke deposition. The outlet reformer composition obtained over Ni–Cu/SiO2 and Rh/Al2O3 catalysts presents a selectivity to hydrogen of approximately 30% with long term stability of the catalysts. This hydrogen rich gas stream can be even improved by WGS and SELOX and it can be considered of high interest to produce electricity for mobile applications by means of fuel cells.Acknowledgements
We are pleased to acknowledge the financial support of the European Commission through contract number ERK6-CT-1999-00012.References
- P. G. Patil, Office of Advanced Automotive Technologies R&D Plan, DOE/ORD/2065, March 1998.
- T. J. Flynn, R. M. Privette, M. A. Perna, K. E. Kneidel, D. L. King and M. Cooper, Soc. Automotive Eng., 1999, 47 Search PubMed.
- J. Patt, D. J. Moon, C. Phillip and L. Thompson, Catal. Lett., 2000, 65, 193 Search PubMed.
- D. J. Moon, K. Screekumar, S. D. Lee and B. G. Lee, Theory and Applications of Chemical Engineering, in: Proceeding of 2000 AIChE Fall Meeting, Pohang University, Korea, 2000, vol. 6, No. 2, 2313 Search PubMed.
- F. J. Mariño, E. G. Cerrella, S. Duhalde, M. Jobbady and M. A. Laborde, Int. J. Hydrogen Energy, 1998, 23, 1095 CrossRef CAS.
- S. Cavallaro, Energy Fuels, 2000, 14, 1195 CrossRef CAS.
- F. Haga, T. Nakijima, K. Yamashita and S. Mishima, React. Kinet.-Catal. Lett., 1998, 63, 253 Search PubMed.
- F. Mariño, M. Jobbagy, G. Baronetti and M. Laborde, Stud. Surf. Sci. Catal., 2000, 130, 2147 Search PubMed.
- S. Matsumoto, Y. Ikeda, H. Suzuki, M. Ogai and N. Miyoshi, Appl. Catal. B: Environmental, 2000, 25, 115 CrossRef CAS.
- V. V. Galvita, G. L. Semin, V. D. Belyaev, V. A. Semikolenov, P. Tsiakaras and V. A. Sobyanin, Appl. Catal A: General, 2001, 220, 123 CrossRef CAS.
- S. Freni, J. Power Sources, 2001, 94, 14 CrossRef CAS.
- J. P. Breen, R. Burch and H. M. Coleman, Appl. Catal B: Environmental, 2002, 39, 65 CrossRef CAS.
- A. N. Fatsikostas, D. I. Kondarides and X. E. Verykios, Chem. Commun., 2001, 851 RSC.
- J. Llorca, N. Homs, J. Sales and P. R. de la Piscina, J. Catal., 2002, 209, 306 Search PubMed.
- V. Recupero, L. Pino, R. Di Leonardo, M. Lagana and G. Maggio, J. Power Sources, 1998, 71, 208 CrossRef CAS.
- S. Velu, K. Suzuki, M. P. Kapoor, F. Ohashi, T. Osaki and N. Miyoshi, Appl. Catal. A: General, 2001, 213, 47 CrossRef CAS.
- S. Ahmed and M. Krumpelt, Int. J. Hydrogen Energy, 2001, 26, 291 CrossRef CAS.
- V. Klouz, V. Fierro, P. Denton, H. Katz, J. P. Lisse, S. Bouvot-Mauduit and Y. C. Mirodatos, J. Power Sources, 2002, 105, 26 CrossRef CAS.
- V. Graziani-Klouz, C. Marquez-Alvarez, C. Mirodatos, F. Michalak, J. P. Lisse, World Pat., WO100320a1, 2000.
- I. Alstrup, M. T. Tavares, C. A. Bernardo, O. Sorensen and J. R. Rostrup-Nielsen, Mater. Corros., 1998, 49, 367 CrossRef CAS.
Footnote |
| † Present Address: Departament d′Enginyeria Quimica. Escola Tècnica Superior d′Enginyeria Química. Universitat Rovira i Virgili. Campus Sescelades 43007 Tarragona España. E-mail: vfierro@etseq.urv.es |
| This journal is © The Royal Society of Chemistry 2003 |