Airborne measurements of peroxy radicals using the PERCA technique
Timothy J. Green*a, Claire E. Reevesa, Neil Brougha, Gavin D. Edwardsb, Paul S. Monksb and Stuart A. Penketta
aSchool of Environmental Science, University of East Anglia, University Plain, Norwich NR4 7DT, UK. E-mail: t.green@uea.ac.uk; Fax: +44 (0)1603 452420
bDepartment of Chemistry, University of Leicester, Leicester LE1 7RH, UK
First published on 12th September 2002
Abstract
The Peroxy Radical Chemical Amplifier (PERCA) technique is a proven method for measurement of ambient levels of peroxy radicals at ground level, but there are no published instances of the technique being used on an aerial platform. Here we describe deployment of a PERCA on the former UK Meteorological Office C-130 Hercules research aircraft. The instrument uses the established method of chemical amplification and conversion of peroxy radicals to nitrogen dioxide (NO2) by doping the sample air-flow matrix with CO and NO, subsequently measuring the NO2 yield with an improved ‘Luminox’ LMA-3 NO2 detector. NO2 from the amplification chemistry is distinguished from other sources of NO2 reaching the detector by periodically injecting CO ∼1 s downstream of the NO injection point (termination mode). Chain lengths (CL's) for the amplification chemistry were typically ∼260 (ground level) to ∼200 (7,000 m). This variation with altitude is less than the variation associated with the ‘age’ of the PFA inlet material where the amplification chemistry occurs; CL's of ∼200 with old tubing to ∼300 with new clean tubing were typical (ground level values). The CL determinations were made in-flight using an onboard calibration unit based on the 254 nm photolysis of 7.5 to 10 parts per billion (by volume, ppbv) of CH3I in air, producing CH3O2 in a quantitative manner. The noise-equivalent detection limit for peroxy radicals (HO2 + RO2) is 2 parts per trillion (by volume, pptv) at 3,650 m when the background ambient ozone levels are stable, based on a 5 min average of five 30 s amplification cycles and five 30 s termination cycles. This detection limit is a function of several factors but is most seriously degraded when there is large variability in the ambient ozone concentration. This paper describes the instrument design, considers its performance and proposes design improvements. It concludes that the performance of an airborne PERCA in the free troposphere can be superior to that of ground-based instruments when similar sampling frequencies are compared.
Introduction
Radicals are fundamental species in tropospheric chemistry: for example the OH radical initiates many tropospheric oxidation pathways, peroxy radicals (e.g. HO2 and RO2) are key reactive intermediates/chain propagators in ozone photochemistry, and the nitrate radical (NO3) is thought to be responsible for the initiation (and propagation) of night-time chemistry.1 The collection and interpretation of free radical measurements under a wide range of atmospheric conditions remains a key issue in tropospheric oxidation chemistry.Considerable progress has been made in measurement of daytime atmospheric radical species in situ at ground level.2–8 Measurements of such radical species from aircraft are less prolific, complicated by a variety of factors such as: sampling issues, variable pressures, size and electrical power considerations and the requirement for fast time resolution. Consequently, measurements of peroxy radical species throughout the troposphere on airborne platforms are still in a stage of development.9–13
Ground-based peroxy radical measurements using the peroxy radical chemical amplification technique (PERCA) pioneered by Cantrell, Stedman and Wendel14 (see also15) have been made extensively by the group at the University of East Anglia (UEA) in the remote marine boundary layer (MBL),8,16–21 and in the free troposphere in the Swiss Alps.22 We report here the deployment of an airborne PERCA based on similar concepts.
The basic PERCA methodology (Fig. 1) of conversion of peroxy radicals (HO2 + RO2) into NO2 and CO2 forms the basis for a relatively simple, lightweight, power-efficient instrument, criteria which are of paramount importance for airborne deployment.
Amplification mode chemistry occurs when both CO and NO are injected at the front of the inlet:
| CH3O2 + NO → NO2 + CH3O | (1) |
| CH3O → CH2O + H | (2) |
| H + O2 + M → HO2 + M | (3) |
| HO2 + NO → NO2 + OH | (4) |
| OH + CO → CO2 + H | (5) |
| H + O2 + M→ HO2 + M | (6) |
| OH, HO2, CH3O2 → wall | (7) |
Termination mode chemistry occurs when CO is injected at the rear of the inlet:
| OH + NO + M → HONO + M | (8) |
| OH, HO2, CH3O2 → wall | (9) |
For a fuller consideration of the PERCA inlet chemistry see Clemitshaw et al.3
The PERCA technique is indirect and so can be subject to interferences, although consideration of these shows they are of lesser magnitude and importance when the technique is used on an aircraft. Other aspects of the technique will also be considered, including the problems associated with sampling the peroxy radicals in a quantitative manner on an aircraft.
This paper details the first airborne deployment of a peroxy radical instrument using the PERCA technique. The instrument described has contributed to the CEC MAXOX (MAXimum Oxidation rates in the free troposphere) and NERC EXPORT (European eXport of Precursors and Ozone by long-Range Transport) science programmes and publication of the details of the instrument will allow critical assessment of these datasets for endusers. Since airborne measurements of peroxy radicals by any technique are costly and complex, it is also hoped our experiences will help others to advance the state of the art in this field.
Experimental procedures
Section 1: The PERCA instrument (PERCA III)
This instrument is the third generation of PERCA deployed by UEA. It shares the same intrinsic chemistry and methods for detection and calibration as the previous two versions,3,22 but differs significantly in the instrument packaging and inlet details–changes that have to some extent been constrained by deployment on an aircraft.Fig. 1 shows a simplified schematic of the instrument that was fitted to the three shelves of a standard mounting system in the front cargo hold of the C-130. The Inlet Unit and Inlet itself were located some 5 m away from the Main Control Unit, mounted above the port side aircraft air-sample pipe (ASP). Lack of space in the ASP area severely constrained the size of the Inlet Unit to 40 × 25 × 18 cm and the inlet mass-flow controllers had to be housed in the Main Control Unit. As a result quite large pressure pulses occurred with each amplification/background valve switch and it typically took 10 s for the LMA-3 to recover from each (see for example Fig. 4). A fourth shelf mounted on a second mounting system carried zero air and nitrogen cylinders. Total electrical power consumption was just under 1 kW and the total instrument weight was ∼150 kg.
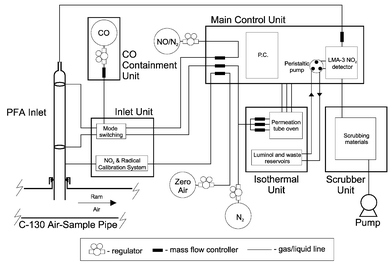 | ||
| Fig. 1 Schematic of PERCA III instrument showing principal components and main interconnections between units. Most gas handling details omitted for clarity, refer to text for details. The initial inlet arrangement, sampling directly from the air-sample pipe, is shown. | ||
The Main Control Unit houses the LMA-3 NO2 detector (LMA-3 Luminox™Scintrex/Unisearch), mass flow controllers (Tylan FC-260′s) and solenoid valves (Galtek 2- and 3-way PFA diaphragm valves, Fluoroware) for controlling instrument gas flows. A Toshiba Tecra 510 CDt laptop computer and ‘Deskstation V+’ docking unit were attached to the top of the Main Control Unit. The Deskstation was fitted with a 16 channel, 16 bit analogue to digital data acquisition card which also carried 21 bits of digital input/output (DAS1602, Computer Boards) and an 8 channel, 16 bit digital to analogue card (DAC 08/16, Computer Boards). These cards used the Computer Boards Universal Library™ and an in-house program written in Microsoft Visual Basic 4.0 to control and log mass-flow controllers, solenoid valves, instrument timing, logging of the LMA-3 raw signal output and logging of other ancillary parameters. A 16 channel multiplexer board (EXP16-RTD, Computer Boards) was later used to extend the number of channels of data logged and the Deskstation/laptop combination replaced with an industrial PC (Wordsworth) housed in the Main Control Unit.
The Inlet Unit housed two 3-way solenoid valves used to switch the inlet reagent flows, a quartz cell, mercury lamp and associated solenoid valves used for making radical and NO2 calibrations. Inlet reagent flows were such that the CO mixing ratio was 7%, and the NO mixing ratio ∼3 ppmv of the 2.0 standard litres per minute (slpm) sampled flow. The PERCA inlet sampled from the port-side air sample pipe (ASP), which is a 2″, unheated, stainless steel pipe carrying turbulent ram air from outside the aircraft boundary layer through the aircraft skin into the cabin space, before passing back out of the cabin using a series of gradual bends. Upstream of the PERCA inlet there were several pre-existing inlets for other instruments which sampled from the core of the flow. The PERCA inlet initially used was a copy of the previously used ground-based Pyrex inlets.3 Pyrex was considered unsafe for aircraft use in such a critical application. It was machined from perfluoroalkoxy Teflon (PFA) and maintained the geometry and spatial separations of the Pyrex inlets used in previous PERCA deployments. The PFA inlet had shown acceptable transmission of radicals when tested alongside a Pyrex inlet during a ground based campaign at the Jungfraujoch observatory in the Swiss Alps (3580 m a.s.l, FREETEX '98). During ground based tests in the laboratory, the PFA inlet had shown a superior chain length, implying less OH wall loss in the amplification region, in agreement with the findings of Jenkin et al.23 With a residence time in the ASP prior to sampling of <0.05 s (based on mass-flow/volume considerations), it seemed likely that enough radicals would be transmitted down the ASP to allow at least a qualitative initial evaluation of the airborne PERCA. Unfortunately as it will be shown later, this did not prove to be the case.
The Isothermal Unit housed three liquid reservoirs in a thermoelectric chiller: luminol (Luminol II solution, Quantitech) to feed the LMA-3 NO2 detector, distilled water for LMA-3 maintenance and waste luminol from the LMA-3. NO2 and CH3I permeation devices were held in a temperature-controlled aluminium block and were used for CH3O2 and NO2 calibrations, detailed later.
The CO Containment vessel housed a 2 l water capacity carbon monoxide cylinder (BOC AC-size) and associated gas handling apparatus, sealing it from the cabin airspace. CO levels inside the box and the aircraft cabin were continuously monitored using commercial hand-held units (Draeger Ltd).
The pump was of the dry rotary vane type (Rietschle VTE-8) pulling 2 slpm through the LMA-3 cell at all altitudes up to 8,000 m. To allow this flow rate up to the maximum C-130 altitude (∼10,000 m), the commercial sample mass-flow controller was replaced with a custom built needle-valve/stepper motor/mass-flow meter combination controlled by the PC software.
PERCA exhaust gases were vented to the rear of the ASP and could be scrubbed to remove the high concentrations of CO and NO present. The exhaust flow (which was damp from contact with the luminol) was first dried with zeolite molecular sieve (BDH, 4A), then passed through Hopcalite (Molecular Products Ltd) which oxidised CO to CO2. Finally the flow passed through a Sofnofil (Molecular Products Ltd) trap which removed the NO and the NO2. CO, NO and NO2 are reduced to low ppmv levels by this method. The Scrubber also contains a charcoal trap to scrub methyl iodide from the permeation source when it is not being used for methyl peroxy calibrations, and an additional Sofnofil trap for cleansing of the NO2 permeation tube outputs when they are not being used.
Zero air (BTCA 178 grade, BOC) and nitrogen (zero-grade BOC, 99.998% pure) were supplied in 10 l water capacity cylinders (BOC F-size) anchored in a specially constructed tray. Both gases were fed through charcoal traps to further purify them. A certified 581 ppm (±1%) mix of NO in nitrogen (Messer) was contained in a small 2 l water capacity cylinder and further purified by passing it through an FeSO4 trap to remove any NO2 present. The CO cylinders were purchased from BOC, and filled as required by Messer with CP grade CO (99.9% purity). An iodinized charcoal trap was used to remove any carbonyl impurities from the CO flow. Two-stage regulators set to 25 psig were used on all gases, (Scott Speciality Gases, model 14 for CO, N2 and zero air, BOC model 51S For the NO/N2 mix), and the gas flows were all regulated with Tylan FC-260 mass-flow controllers which were regularly calibrated with bubble flow-meters.
Section 2: Detector system and calibrations
The principle of operation of the LMA-3 NO2 detector is described thoroughly in Kelly et al.24 When used at reduced pressure, the luminol feed to the LMA-3 cells was often disrupted by degassing and at high altitudes, the disruptions are almost continuous. With use of the very basic ‘upgraded’ peristaltic pump available as an accessory from Unisearch, wide bore gas and liquid tubing, replacement fittings for liquid and air lines to the cell, and vacuum-testing to ensure the assembly was sealed, the LMA-3 performed comparatively well up to 6,000 m.An additional complication arises because the PERCA technique measures the modulated NO2 from the amplification chemistry on top of a NO2 background from ambient NO2, ambient ozone (that is titrated to NO2 by the high mixing ratios of NO used in the inlet) and thermal decomposition of NO2 reservoir species. Forward motion of the aircraft, especially in regions where airmass mixing is occurring, make the ambient O3 fluctuations particularly problematic and can swamp, subtract or add to the signal from the amplification chemistry. The only reliable method to overcome this problem is to run a second detector and inlet to determine the contribution that other NO2 sources are making to the LMA-3 signal.2 The initial PERCA installation on the aircraft was motivated by the need to probe the radical concentrations of clean, well-mixed air masses, in which rapidly fluctuating ozone is not observed. In this type of deployment single channel instruments can still provide excellent data.18
The LMA-3 detector is calibrated using a wafer permeation tube (VICI Metronics) which produces 5.7 ppm of NO2 in 50 standard cubic centimeters per minute (sccm) of zero-grade N2 (mass-flow controlled). The permeation tube is kept in an insulated aluminium block at 40 ± 0.2 °C with a PID temperature controller (Omega CN76000) and K-type thermocouple controlling a thermofoil heater element (Minco). This block also held the CH3I permeation tube and an additional NO2 tube (see later in the text) and was housed in the Isothermal Unit, which was removed from the aircraft after each flight and kept powered on the ground to ensure that the permeation tube outputs stayed constant. The permeation tube output rates were found by measuring the weight loss with time while keeping the permeation tubes at 40 °C in the Isothermal Unit for several weeks (Table 1). Further dilution of the NO2 calibration flow with 3.5–5 slpm of BTCA 178 zero air (BOC) resulted in an overflow of 46–75 ppbv NO2 which could be added to the inlet via a sidearm. Two point calibrations and an interpolated zero were performed and the LMA-3 sensitivity determined from a plot of mixing ratio versus NO2 response (Table 2 shows typical values for a flight).
| Tube | Permeation rate/ng min−1 |
|---|---|
| CH3I calibration | 251.4 ± 9.6 |
| NO2 calibration | 495.9 ± 12.8 |
| NO2 linearisation | 175.2 ± 17.0 |
| Altitude/m | NO2 sensitivity/V molecule−1cm3 | CL |
|---|---|---|
| 0 | 5.66 ± (0.11) × 10−13 | 281 ± 18 |
| 4880 | 7.27 ± (0.11) × 10−13 | 277 ± 15 |
| 6100 | 6.89 ± (0.42) × 10−13 | 266 ± 7 |
| 30 | 5.23 ± (0.44) × 10−13 | 291 ± 14 |
| 2130 | 6.47 ± (0.25) × 10−13 | 321 ± 14 |
| 3960 | 6.66 ± (0.06) × 10−13 | 299 ± 12 |
| 5670 | 6.79 ± (0.11) × 10−13 | 291 ± 30 |
| 5490 | 7.33 ± (0.17) × 10−13 | 288 ± 9 |
| 2590 | 6.21 ± (0.13) × 10−13 | 319 ± 7 |
| 1980 | 6.12 ± (0.22) × 10−13 | 322 ± 18 |
| 300 | 5.73 ± (0.27) × 10−13 | 286 ± 15 |
| 5490 | 7.89 ± (0.21) × 10−13 | 255 ± 2 |
| 4880 | 7.15 ± (0.19) × 10−13 | 290 ± 31 |
| 0 | 5.30 ± (0.11) × 10−13 | 289 ± 17 |
A constant offset of 39 ppbv NO2, in 50 sccm N2 could be generated in the same manner as the calibration flow. This ‘linearisation flow’ was added to the sample matrix if sampled NO2 levels were so low that the LMA-3 signal fell into the non-linear region of the detector.24 The linearisation flow was required for chain length (CL) calibrations, which were done in zero air. It was usually kept flowing to the LMA-3 during measurement mode too, as the constant offset provided a useful check of detector performance.
Methyl peroxy radicals for determining the amplification CL of the instrument were produced using the permeation tube of methyl iodide (VICI Metronics) to generate a flow of 0.8 ppm methyl iodide in 50 sccm of N2, diluted again in 3.5–5 slpm BTCA 178 zero air. The resulting blend was passed through an 80 cm3 ‘Suprasil-B’ cylindrical cell, and illuminated using an 8 W germicidal mercury lamp (Starna Industries) with a strong output at 254 nm. The CH3O2 concentration produced by the cell is given by:3
| [CH3O2]cal = jCH3I[CH3I]tres | (10) |
| tres = the residence time in the cell | (11) |
| jCH3I = the photolysis rate of methyl iodide in the cell | (12) |
The photolysis rate was measured in the laboratory under static conditions using elevated concentrations of CH3I and conventional UV spectroscopy as described in Clemitshaw et al.3 The calibration system fed a mixture of between 35 and 80 pptv of radicals in dry zero air to the PERCA inlet system during flight and on the ground. A short length of PFA tubing and one PFA solenoid valve allowed the output of the cell to be connected to the PERCA inlet sidearm with <1 pptv loss of CH3O2. The amount of NO2 produced from the difference between the average of ∼20 s of amplification mode data and ∼20 s of termination mode data, Δ[NO2], can be related to the dry air CL:
| Δ[NO2] = CL × [CH3O2]cal | (13) |
The detector and NO2 calibration system were initially characterised during the TACIA and ACSOE campaigns, flown in the UK and the Azores from August 15th to September 23rd 1997. The NO2 levels experienced were usually below the 50 pptv manufacturer's detection limit for the LMA-3 and the signal from the instrument was virtually entirely due to the presence of ozone, which is a known source of interference for the detector. The high quality peristaltic pump initially installed and previously used for ground-based deployment was discarded in favour of the simpler ‘upgraded’ peristaltic pump (available from Unisearch as an accessory) set to deliver luminol at ∼1 ml min−1. Ambient ozone data was scaled to calculate the expected ozone interference to the LMA-3 ‘NO2’ signal (as shown in Fig. 2), using a response equivalent to 0.01 ppbv NO2 per ppbv of O3, which is in line with the manufacturer's quoted 1% interference from O3 at low NO2 levels (<5 ppb). During Flight A577, O3 measured directly by the aircraft TECO 49 ozone analyser, varied from a minimum of 20 ppbv in the MBL to a maximum of 90 ppb at the upper flight levels. The real NO2 levels measured by the UEA NOxy instrument NO2 channel reached a maximum of 300 pptv. It is clear from Fig. 2 that virtually all of the LMA-3 signal is due to the interference of O3. The small deviations between the two traces probably result from the need for altitude and PAN interference corrections when making quantitative measurements of ambient mixing ratios of NO2 using an LMA-3 detector.24Fig. 2 demonstrates the quantitative airborne response of the detector system using O3 as a proxy for NO2. When the PERCA amplification chemistry is operating, NO2 levels are well above the LMA-3 detection limit (∼3 ppbv NO2 from the amplification chemistry, superimposed on a background of 20 to 100 ppbv of NO2 from titration of ambient O3 by the high NO mixing ratios in the inlet).
 | ||
| Fig. 2 Plot of calculated O3 interference (grey) and LMA-3 signal (black) from TACIA flight A577. | ||
Discrimination of this small level of amplified NO2 can only be precisely achieved if the background ozone varies by a small amount between each termination measurement of the PERCA. The standard deviation from 20 s of an optimum LMA-3 signal is 0.18 ppbv or ∼1 pptv radical equivalent. This level essentially represents the noise level intrinsic to measurements using the LMA-3 and the ultimate detection limit of the PERCA. The stability of background ozone levels during the MAXOX campaign is evidenced by typical standard deviations (20 s of termination signal) of around 0.34 ppbv (or ∼2 pptv radical equivalent). By using the moving average of two termination measurements, the precision associated with the amplified NO2 can be improved to ∼0.25 ppbv, which is less than 10% error when the amplified NO2 is 3 ppbv. Signal averaging can be used to refine the precision, but is of limited use on these timescales on an aircraft. Ozone data from the aircraft can be used to correct a PERCA signal (or to flag imprecise data) only when the fluctuations are greater than the 1 ppbv precision of the ozone instrument, equivalent to ∼33% error. Between ozone variations of ∼1 to 0.2 ppbv, standard deviations of the amplified and background signal provide a measure of data quality.
Section 3: Characterisation of the airborne PERCA
The full PERCA instrument was tested on 3 flights during July and August 1998, having added the PFA PERCA inlet, radical calibration system, NO/N2 reagent gas bottle and the CO reagent containment system. Airborne calibrations were successful but the optimal instrument detection limit was estimated to be around 10 pptv of radicals at 4,000 m and no ambient radicals could be observed. It was concluded that the inlet system was unsatisfactory. The comparatively poor detection limit of the instrument at this time was due to a systematic offset of the amplification signal from the termination signal, caused by the inlet design relying on 1″ diameter O-rings to seal the toroidal injectors against the differential between cabin pressure and ambient pressure. This deficiency, combined with the likelihood of some losses down the stainless steel ASP, account for lack of ambient detection of radicals in this flying period.In an attempt to resolve the problems with the original PFA inlet, a small component of Flight A694 on the 14th July 1999, between 11:45 and 13:15 at FL145 (∼4,400 m) was used to compare the original PFA inlet with two alternatives (Figs 3a and 3b). Inlet #1 was the original aircraft inlet but connected to a pre-inlet manifold of 3/8″ o.d. PFA tube that lined the inside of the port-side ASP, ducting unimpeded ram air into the cabin solely for sampling with the PERCA. Inlet #1 had previously used a 1″ ‘Cajon’ fitting (SS-16-UT-A-20-BT, Swagelock) welded to the ASP (see Fig. 1). The catheter was sealed within this ‘Cajon’ fitting using a bored-through 3/8″ to 3/4″ pipe weld connector (SS-600-1-12W, Swagelock). The ram air passing down the pre-inlet manifold was returned to the rear of the ASP after a ‘T-piece’ connection to the PERCA inlet (shown in Fig. 3a for Inlet #2).
 | ||
| Fig. 3 (a) The configuration of Inlet #2 which sampled from the same location as Inlet #1, the port air-sample pipe. This diagram shows the heater which was installed post A694 for the MAXOX flights. (b) shows the configuration of Inlet #3 which had an optimal sampling of air-flow, but was located some distance from the rest of the PERCA instrument on the starboard side of the aircraft. | ||
Inlet #2 was constructed from PFA tubing, with the reagent addition achieved by injection at two catheterised t-pieces (‘Galtek’, Fluoroware) located 1.0 m apart (∼1 s residence time at 2 l min−1 at atmospheric pressure, i.e. the same as Inlet #1). It was swapped in-flight with Inlet #1 so it was fed with the same ram air from the 3/8″ PFA ASP manifold. Inlet #3 was constructed from ¼″ PFA in the same manner as Inlet #2 but using a longer length of tubing to maintain the same residence time and utilising a skin penetration and ‘Rosemount’ inlet on the starboard side of the aircraft normally used by the UEA NOxy instrument. This allowed direct sampling of the outside air-flow, albeit through the small ‘Rosemount’ section of stainless steel, to give a comparison of the losses associated with Inlets #1 and #2 and the pre-inlet manifold.
The comparable data relating to each inlet are shown in Table 3. All three inlets showed very similar sensitivity to NO2, the error quoted being the standard error in the slope of the regression line from a three-point calibration. The three values are within the errors in the measurements and the estimated uncertainty of 5% for the NO2 calibration of the instrument. The length of tubing required for Inlet #3 resulted in a small pressure drop while a small pressure rise was caused by the ram air effect of the PFA ASP catheterisation used for Inlets #1 and #2. The CL was much lower for Inlet #1, and similar for Inlets #2 and #3. We have found in laboratory tests that the use of a longer, narrower inlet reactor gives superior CL's compared to the ‘classic’ inlet geometry used in previous ground based studies.3,22 The reason for this is almost certainly improved mixing; the catheterisation employed in Inlets #2 and #3 allows reagent injection into the centre of the sampled flow where the flow velocity is fastest (all configurations should exhibit linear plug flow based on their calculated Reynolds’ number). By the time flight A694 took place in July 1999 the original PFA aircraft inlet (Inlet #1) had been deployed on 12 flights totalling ∼57 hours flying time over a one year period and the CL had been deteriorating progressively. Some ‘ageing’ has been observed with all the PFA inlets used since. It may be due to loss of the smooth PFA surface as aerosol and dust builds up on it, the propensity for PFA to carry a static charge aiding the process. Cleaning via usual methods (ultrasonic bath combined with solvents/detergent) does not regenerate the CL and so now all inlet PFA is replaced just prior to take-off. Pre- and post-flight ground calibrations produce CL's that agree within the accuracy of the instrument (40%), supporting the fact that the aging is a gradual process and not significant during a single flight. Regular CL calibrations during flight show that CL's vary as a function of altitude in a reproducible manner, with no sudden degradations after sampling polluted air masses. No tests have yet been undertaken on the effect of sampling air masses with significant precipitation taking place. Inlet #2 and #3 had been constructed and tested thoroughly on the ground prior to this flight which is the probable explanation for their CL’s being lower than those obtained for brand new tubing (e.g. see values in Table 2).
Inlets #2 and #3 produced clear modulations in the NO2 signal from ambient air (see Fig. 4), but Inlet #3 showed the larger mixing ratio of sampled radicals. This is consistent with some loss of radicals down the PFA pre-inlet manifold which catheterised the ASP. Inlet #1 showed no modulations when ambient air was sampled, the uncertainty due to the poor S/N on the modulations being greater than the levels of radicals present based on the data from Inlets #2 and #3.
The switching time, the time taken for the detector signal to stabilise after a switch from amplification to background mode or vice versa, was quickest for Inlet #2 (10 s), and slowest for Inlet #3 (13 s). This is consistent with the longer length of tubing allowing diffusive blurring of the switch. Inlet #3 also had the reagent MFC's located furthest from the inlet since the ‘Rosemount’ used was mounted on the opposite side of the aircraft to the instrument rack.
At least 46% radical loss appears to have occurred in the PFA Inlet #2 (compared to Inlet #3, see Table 3). The low CL and poor definition of the amplification and background modes with Inlet #1 made it impossible to see ambient mixing ratios. Direct sampling of the airflow was clearly desirable (Inlet #3) but not available during the normal photochemistry instrument fit to the C-130. The verification of the poor performance of Inlet #1 confirmed why previous flights had failed to observe ambient radicals.
For the MAXOX campaign (July 23rd to August 11th, 1999) a modified version of Inlet #2 was used, having a spring clip to hold the forward length of PFA secure within the ASP, and a heater capable of keeping the amplification zone of the inlet at a constant 30.0 ± 1.5 °C. The PFA pre-inlet manifold lining the ASP could not be heated and was thus at ambient air temperature. This inlet produced the first conclusive modulations over a variety of flight regimes. For these and subsequent flights, all PFA exposed to peroxy radicals was replaced prior to each flight to avoid degrading CL's.
During the MAXOX campaign, the in-flight calibration facility for methyl peroxy radicals allowed dry air CL's to be determined for flight levels ranging from 15 m to 7,000 m. Some 60 hours of flying were made during the summer MAXOX flights. The scientific objective was to determine the chemical conditions for maximum oxidation of various trace gases, and to determine the maximum production rate of ozone with respect to season. Flight profiles were mostly designed to optimise the PERCA performance: long level runs, with NO2 and CL calibrations performed on each run. Air masses were chosen where background ozone levels were not fluctuating significantly on a timescale faster than the 1 min required for a PERCA measurement cycle.
Section 4 : Laboratory characterisation of potential radical losses within the aircraft inlet
The first order wall loss of peroxy radicals in zero air down ¼″ PFA tube as a function of temperature was determined in the laboratory to confirm the observations made with the various inlet systems on the aircraft. The CH3O2 loss fitted an exponential decay versus temperature and first order loss rates of between 0.2 s−1 at 0 °C to 0.8 s−1 at −80 °C were obtained. Modelling of the system with the FACSIMILE package25 indicates self-association reactions will account for <1.5% of loss at −80 °C. Thus better than 96% transmission of methyl peroxy radicals through the aircraft inlet system was expected under worst-case flight conditions (residence time in the unheated, PFA pre-inlet manifold of 0.04 s from mass-balance considerations; outside air temperature of 223 K).The only source of HO2 available was photolysis of water vapour, but condensation occurred at reduced temperatures because of the high relative humidity (near 100% at room temperature) used in the photolysis cell. At room temperature (22 °C), 36% loss of HO2 occurred down a 30 cm length of clean 1/4″ od PFA tube. At −5 °C 87% loss occurred down the same piece of tube, but rapid condensation of water resulted in 100% loss of the HO2. If the tube was allowed to warm to 22 °C again, HO2 transmission recovered to approach the previously observed levels. From these experiments it can be concluded that while good transmission of methyl peroxy should occur down the PFA aircraft inlet, there could be significant losses associated with HO2 (especially in condensing conditions), consistent with the difference in ambient levels measured with Inlet #2 and Inlet #3 on flight A694.
A number of other readily available materials were tested for transmission of methyl peroxy radicals. All measurements were made at 22 °C. There was no detectable loss of CH3O2 down 30.5 cm of PFA (internal volume = 3.7 cm3), while the same length of nickel tube (internal volume = 6.0 cm3) resulted in 61% loss. The same length of 316 stainless steel (internal volume = 6.0 cm3) resulted in an 81% loss, and a reduction in the total amount of NO2 reaching the detector. A 15 cm length of PFA tube (internal volume = 1.8 cm3) resulted in no detectable loss at 22 °C but the same length of high purity gold tubing (internal volume =2.9 cm3) resulted in 47% loss. From these experiments we concluded that clean PFA was considerably better than stainless steel, gold and nickel for radical transmission, although it was unfortunate that we could not compare the same lengths and internal volumes directly to derive more quantitative data. A more thorough testing process is planned for the next generation PERCA inlet since from an engineering viewpoint, neither glass nor PFA are particularly convenient materials to work with. Fabrication of an inlet from a suitable metal would have significant advantages for mounting on an aircraft.
Discussion: measurements of peroxy radicals using PERCA III
PERCA III in its latest form has been used in three campaigns: the MAXOX summer experiments which took place in July/August 1999, the NERC UTLS ACTO (Atmospheric Chemistry and Transport of Ozone) experiments which took place in April/May 2000, and the NERC EXPORT experiment which took place in July/August 2000. A major objective in all these experiments was to combine measurements of peroxy radicals with measurements of other parameters (including ozone, nitric oxide (NO), the photolysis rate of ozone (j(O1D)), and the water vapour concentration), to determine in situ production and loss rates for ozone in different air masses and at different times of the year. It is not the purpose of this paper to discuss the results in detail but more to present data that indicates the performance of the instrument and the usefulness of the data for achieving the objectives of the experiments.An example of the raw data output from PERCA III is shown in Fig. 4 for flight A704 on 9 August 1999. The signal from the LMA-3 is shown as a thin black line as the chemical amplifier is switched from the amplification to termination mode. The radical signal is the difference between the two interpolated grey lines (expressed as an NO2 concentration) divided by the CL, which is regularly determined in flight using the on-board calibrator. It can be seen that the signal does not vary much over the ten minutes shown, apart from a short period between 13:28 and 13:30 hours, where a disturbance in the background signal is caused by changes in the ambient ozone concentration (ozone reacts with NO in the inlet to produce NO2, thus altering the background level). The regular abrupt changes in the raw signal are caused by discontinuities in the reagent flows when the chemical amplifier is switched from amplification to termination mode and vice versa. These sections of the data are removed before determining the averages shown in Fig. 4.
 | ||
| Fig. 4 Raw LMA-3 signal from MAXOX flight A704 at 5500 m. Amplification averages shown as open triangles, termination mode averages as open squares. Signal corresponds to approximately 8 pptv radicals. | ||
The full dataset for peroxy radicals obtained on flight A704 can be seen in Fig. 5, the peroxy radicals measured by PERCA III on this flight varied from close to zero to about 12 pptv. The measured peroxy radicals have been compared with values calculated with a box model26 which uses constrained input of ozone, j(O1D), j(NO2) H2O, NO, CO and peroxides, and assumes a methane concentration of 1.8 ppmv. The radical levels predicted by the model are also shown in Fig. 5 with values calculated for CH3O2 being of similar magnitude to those measured by the PERCA. The temporal variation in the measured data is also correlated quite well with the modelled CH3O2 values. The PERCA data at the lowest flight levels is undoubtedly low due to the water vapour effect suppressing the CL. Elsewhere Fig. 5 suggests strongly that the airborne PERCA with its rudimentary inlet system (Fig. 3a) is not measuring HO2 radicals quantitatively. This observation is in agreement with the findings of the laboratory tests that showed that CH3O2 radicals are transmitted well through even cold PFA tubing but that losses of HO2 were high. The theory that HO2 radicals are preferentially lost on the PFA pre-inlet is supported by the limited tests carried out on the aircraft with different inlet configurations. Table 3 shows that the signal almost doubled with Inlet #3 which sampled essentially unimpeded ambient air. The modelled CH3O2 values are consistently slightly lower than the PERCA data, although the agreement is well within the instrumental errors. Allowing 10% transmission of HO2 down the pre-inlet would bring the measured and modelled radical levels into slightly better agreement.
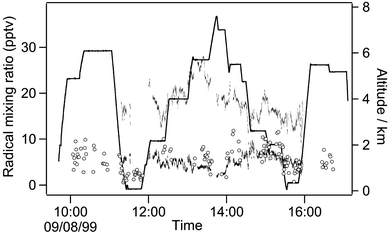 | ||
| Fig. 5 PERCA III signal (open circles) from MAXOX flight A704 compared with modelled CH3O2 data (black dots) and modelled HO2 data (grey dots). Altitude is shown as a black line. | ||
Fig. 6 shows the radical data from all MAXOX flights plotted as a function of altitude with no filtering criteria applied. This figure indicates that the PERCA is consistently measuring low levels of peroxy radicals at the highest altitudes (typically around 5 pptv or less above 5,500 m and always below 15 pptv). By contrast the modelled values for HO2 for the MAXOX data above 4 km altitude are very seldom less than 10 pptv, except in high NO conditions, and more often above 15 pptv.
 | ||
| Fig. 6 Plot of PERCA III signal as a function of altitude for all MAXOX data. | ||
Significant levels of peroxy radicals (HO2 + RO2) between 4 and 6 km have been measured by Reiner et al.,10 (10–20 pptv on two flights over south Germany). Brune et al.9 have observed greater than 12 pptv of HO2 in clear air over North America at similar altitudes. The low (i.e. close to detection limit) PERCA radical signals seen at high altitudes during all airborne deployments further strengthens the theory that PERCA III is insensitive to HO2. Further analysis is required, but the conclusion drawn from the data considered so far, is that PERCA III measures organic peroxy radicals quantitatively. It is unlikely to detect HO2 because of losses in the simple inlet system used to date.
At the end of the MAXOX campaign on 11 August 1999 a complete eclipse of the sun was experienced in the southern UK.27 The opportunity was taken to test the performance of various instruments used for studies of atmospheric photochemistry on the C-130 aircraft. These included radiometers,28 the 4-channel NOxy instrument29 and PERCA III. Fig. 7 shows the variation with time of 3 instruments on the aircraft during the eclipse period. The aircraft was flying in a rather invariant ozone field (O3 ∼ 50 ppbv), at a constant altitude of 2,100 m, around 50 °N, 15 °W. The exact aircraft positioning was chosen to be cloud-free and to optimise the PERCA signal with respect to altitude and to background signal variation.
 | ||
| Fig. 7 PERCA III data, (open circles, 1 min averages), NO data (black dots, 10 s averages), j(O1D) levels (black line,1 s data) and altitude (grey line) during the eclipse, Flight A705. | ||
The time of the eclipse and achievement of total darkness is shown primarily by the j(O1D) trace in Fig. 7; an observer using the aircraft sextant observed 47 s of totality beginning at 09:59:37 GMT. The NO signal from the NOxy instrument follows the j(O1D) variation precisely through the decline and rise in light levels (NO is produced by photolysis of ambient NO2). Concentrations vary from an average of 8 pptv down to zero, with a recovery to approximately 15 pptv following the eclipse period. The PERCA signal shows more variance than the NO signal, but it is quite possible to discern the decay as the light levels were declining, and the rise when light levels returned to normal around 11:00 hours GMT. CH3O2 is clearly related to j(O1D) although the data quality is insufficient to allow us to determine whether there is a first order or a square root relationship between these two quantities. In clean air with NO less than 50 pptv it would be expected that CH3O2 is proportional to √j(O1D) as was observed at Cape Grim in 1995.16,18,19 The substantial fall in the RO2 and NO concentration later in the flight around 11:30 GMT is associated with an air mass change at the extreme edge of the operational area.
It has been observed that water vapour suppresses the PERCA radical response.6,30,31 Since the PERCA CL is determined in dry (H2O < 5 vppm) bottled air, the discrepancy between the calibrated dry-air chain length, and that expected when operating in moist ambient air, can be very large–up to 70%.8 Fortunately, deployment of an aircraft instrument means that any such correction is most significant at the lowest flight levels, which constitute a relatively small component of the flight data. Above 3,500 m the correction will be less than 10–15% (c.f. Zanis et al.22) but below this altitude application of some sort of correction factor (or calibration at ambient humidity) should be applied. The consistently low radical levels at low altitudes shown in Figs 5 and 6 are at least partially due to the lack of any correction factor being applied to the data, although elevated NO levels also suppress peroxy radical levels. Salisbury et al.,8 and Burkert et al.,32 have both shown that it is possible to correct PERCA data for the water vapour suppression of the CL with some confidence. It is intended to verify that a similar suppression is applicable to the MAXOX type inlet.
While the PERCA as deployed so far clearly does not measure the sum of HO2 + RO2 quantitatively, it does appear to provide a reliable estimate of CH3O2. The accuracy should be ∼35% based on the estimated errors associated with the Cl,3 if the additional error associated with the effect of humidity on the CL is ignored. Further work is required to be sure that all HO2 is quantitatively lost on the pre-inlet, but the modelled and measured values for the MAXOX flights indicate agreement for CH3O2 to within 50% for the majority of the dataset. This agreement is remarkable, but consistent with a total estimated accuracy of 40% for the instrument in its present form (at altitudes above 3,500 m) with a 1 min detection limit for CH3O2 of 2 pptv.
Since the technique is an indirect one, PERCA radical measurements could have a contribution from radical reservoir species. The most likely interferants in the troposphere are peroxy nitric acid (PNA), HO2NO2, and peroxy acetyl nitrate (PAN), CH3C(O)O2NO2. Both PNA and PAN are thermally labile radical reservoir species and any decomposition that produces a radical fragment within the PERCA sampling system, (prior to the termination chemistry occurring) can result in a spurious additional radical signal. Since the PERCA is modulated between amplification and termination modes, only radical species produced before the second injector (more accurately before the CO from the termination injector mixes with the bulk flow) will be observed as interference, since after this point any additional NO2 formation is present in both the amplification and termination modes. Similarly, any direct NO2 production within the pre-inlet manifold from decomposition will appear in both modes and the amplified levels of NO2 from the CL chemistry, determined from the difference between the two modes, will not be affected. The small quantities of NO2 produced in this manner are negligible compared to the amounts produced from ambient O3 titration. High levels of NO2 can reduce the CL, FACSIMILE modelling indicates that with a CL = 315 ∼80 ppbv of sampled O3 will suppress the CL by 12% the effect being progressively less for lower CL's. Thus PAN decomposition should not affect the airborne PERCA, but operation in regions where large O3 concentrations are sampled would require some care in applying a valid CL.
The PNA decomposition rate constant is some 200 times faster than that for PAN based on recommended rate data for T = 300 K33 and with PNA levels of ∼5 to 150 pptv postulated in the cold free troposphere, PNA could be an interference in PERCA measurements. In fact, any HO2 measurement technique that disturbs the thermal equilibrium of the sample matrix could be subject to such interference. Work by Zanis et al.,22 has shown that free-tropospheric PNA levels of between 8 and 66 pptv would be expected, associated with observed NO mixing ratios of between 10 pptv and 117 pptv respectively. Observed MAXOX NO levels rarely exceeded 80 pptv, so a ‘worst’ case for PNA interference of 60 pptv of PNA can be assumed for an altitude of 3,500 m (pressure ∼ 490 Torr). Under these conditions the residence time in the heated zone of the PERCA would be no more than 0.8 s. Assuming that the bulk temperature of the gas in this region attains the wall temperature instantly, calculations show that 5% of the PNA would decompose, producing 3 pptv of radicals. As the altitude increases, colder temperatures favour sequestration of HO2 as PNA (assuming sufficient NO2 is present). The interference of these higher amounts of PNA is moderated by reducing residence times in the PERCA inlet, caused by the greater volume of the mass-flow controlled sample flow. At lower altitudes, a longer residence time would allow a greater fraction of the sampled PNA to decompose, but the increasing ambient temperature means that ambient PNA formation is increasingly unfavourable. Since the true average sample flow temperature in the heated zone must be less than the wall temperature, and the temperature must increase gradually as the flow passes down the inlet towards the termination injection point, the true interference of PNA is likely to be insignificant.
As part of the development work for a future airborne instrument, a dual-channel PERCA instrument was operated at the Jungfraujoch (3580 m a.s.l, for site details see Zanis et al.34) in early spring. One inlet was held at 30 °C and the other heated to 40 or 60 °C. No detectable enhancement was observed in the radical signal with the hotter inlet, placing an upper limit on PNA concentrations at the Jungfraujoch at this time of 10 to 20 pptv. Dual-channel airborne instrumentation should allow assessment of the interference of PNA on radical measurements over a wider range of tropospheric altitudes and NOx mixing ratios. Reliable measurements of PNA are also desirable to aid in understanding ozone production efficiencies in the free troposphere.35
In view of the experimental versatility, and improved background signal retrieval, the next generations of airborne PERCA will be dual-channel. During ACTO (May 2000) and EXPORT (July and August 2000) a second detector and a simple extra inlet sampling ambient air doped with 3 ppmv NO was added to PERCA III. Its purpose was to provide a continuous measurement of the ambient ozone interference to the PERCA and it yielded promising signs that a second channel can extend the range over which optimal operation of the PERCA is possible. Deployment of a full dual-channel PERCA (as opposed to the single-channel PERCA with additional O3 background channel) on the ground has yielded greatly improved precision in what would ordinarily be sub-optimal conditions for a single channel instrument. With the use of two full PERCA channels, data can be recorded continuously, switching less frequently between amplification and background modes. The two channels are operated out of phase to acquire background data from one channel while the other channel is amplifying and vice versa.
Conclusions
This paper has detailed the implementation of a calibrated PERCA instrument that can be deployed on an aircraft. While this instrument performs satisfactorily under optimum conditions of stable ambient ozone in the dry free troposphere, there are still a number of concerns that must be addressed before the data from this PERCA can be considered quantitative. The first concern is one that applies to all measurements of reactive species: that of quantitative sampling, in our case, of ambient peroxy radicals. Modelling, coupled to laboratory studies of the wall losses of methyl peroxy radicals and HO2, give confidence that the PERCA inlet as deployed on the C-130 measures only methyl peroxy radicals in the free troposphere. With an inlet designed to minimise surface losses, the PERCA should measure the sum of organic and inorganic peroxy radicals (RO2 + HO2) successfully.Atmospheric fluctuations in ozone are the single largest factor affecting PERCA precision. The total uncertainty in an airborne PERCA operating in optimal conditions is around 40%, when ozone levels are stable, and then the precision of the airborne signals surpass the performance seen in such places as Cape Grim.18 However, when ozone levels change rapidly, such as in air-mass mixing regimes in the UT/LS region, it is impossible to extract any meaningful data using the PERCA technique without a second inlet (see also Cantrell et al.2).
The suppression of the dry air CL by water vapour remains the largest source of uncertainty in the accuracy of the PERCA technique. Nonetheless, for an aircraft instrument, and especially one deployed in the upper troposphere, the water vapour effect is much less important (less than 15% above ∼3500 m). A means of accurately compensating for the effect is essential if reliable profile data from a PERCA is to be obtained into the MBL, where the correction can be up to 70%.8
Prompted by a thorough modelling and laboratory programme,23 and flight tests of the PERCA, improvements to the NO2 detection system have resulted in a relatively trouble-free method of detecting the amplified NO2 from the PERCA using the modified LMA-3 detector. A problem highlighted in the modelling studies; namely low predicted CL's at altitude, has been found to be compensated for by improved inlet reagent mixing, allowing the PERCA technique to be applied to the bulk of the troposphere. Operation of a PERCA inlet at a reduced pressure may provide a useful way around many of the complications encountered with an ambient pressure inlet (e.g. varying NO2 sensitivity, varying CL, and the loss of measurement time to make the necessary calibrations), if the additional complexity of implementation was considered acceptable. A constant flow profile down the inlet should result in much better characterisation of inlet losses and processes. It will be noted however, that since the PERCA III CL and NO2 calibrations are undertaken at the prevailing ambient pressure of the flight level, all pressure effects should be accounted for. A reduced pressure inlet would mainly serve to reduce in-flight calibration time, so providing additional measurement time.
In spite of some deficiencies, the PERCA principle of chemical amplification and conversion of radicals to NO2 can be applied successfully in an airborne environment. With refinement the technique could provide a low cost, relatively simple, means of providing in situ radical data from aircraft. It could be applied throughout much of the troposphere, complementing existing methods of measuring OH and contrasting with the few existing methods of detecting RO2 and HO2.
Acknowledgements
We would like to thank the following bodies and individuals:The technicians of the UEA mechanical and electrical workshops, especially Mr. Frank Robinson and Mr. Roger Humphrey, who often endured highly pressurised workloads to meet the tight deadlines. The UK Meteorological Office for their help with the installation of the PERCA and the unfunded ‘piggy-back’ test flights on to the C-130, noting especially the patience and assistance of Mr. Joss Kent and Mr. Ken Dewey. Dr. Mike Jenkin, Dr. Samantha Ashbourn and Dr. Kevin Clemitshaw for helpful discussions during and after the reduced pressure laboratory tests and modelling work. The Natural Environmental Research Council – Atmospheric Research Airborne Support Facility for providing funds to cover test-flight A694. Dr. Hannah Barjat from NERC, for modelling and planning work associated with the eclipse flight A705.
The PERCA aircraft installation was funded through NERC ACSOE grant GST/02/1263 and j(O1D) data through GR9/102743. ACSOE flying was funded through ACSOE Core programme grant number DST/03/02/07/01, TACIA flying through CEC Grant number ENV4-CT95-0038. MAXOX was funded by CEC grant number ENV4-CT97-0525, ACTO by NERC UT/LS Ozone grant GST/02/2466 and EXPORT by NERC grant GR3/13095. This work is ACSOE paper number: ACP173.
References
- Atmospheric Chemistry and Global Change, ed. G. P. Brasseur, J. J. Orlando and G. S. Tyndall, Oxford University Press, Oxford, 1999 Search PubMed.
- C. A. Cantrell, R. E. Shetter and J. G. Calvert, Anal. Chem., 1996, 68, 4194 CrossRef CAS.
- K. C. Clemitshaw, L. J. Carpenter, S. A. Penkett and M. E. Jenkin, J. Geophys. Res., 1997, 102(D21), 25–405 CrossRef.
- D. J. Creasey, P. A. Halford-Maw, D. E. Heard, M. J. Pilling and B. J. Whitaker, J. Chem. Soc. Faraday Trans., 1997, 93, 2907 RSC.
- D. J. Tanner, A. Jefferson and F. L. Eisele, J. Geophys. Res., 1997, 102(D5), 6415 CrossRef CAS.
- C. M. Mihele and D. R. Hastie, J. Atmos. Oceanic Technol., 2000, 17, 788 Search PubMed.
- M. Hanke, J. Uecker, T. Reiner and F. Arnold, Int. J. Mass Spect., 2002, 213, 91 Search PubMed.
- G. Salisbury, P. S. Monks, S. Bauguitte, B. J. Bandy and S. A. Penkett, J. Atmos. Chem, 2002, 41, 163 CrossRef CAS.
- W. H. Brune, I. C. Faloona, D. Tan, A. J. Weinheimer, T. Campos, B. A. Ridley, S. A. Vay, J. E. Collins, G. W. Sachse, L. Jaeglé and D. J. Jacob, Geophys. Res. Lett., 1998, 25, 1701 CrossRef CAS.
- T. Reiner, M. Hanke, F. Arnold, H. Ziereis, H. Schlager and W. Junkermann, J. Geophys. Res., 1999, 104(D15), 18647 CrossRef CAS.
- I. Faloona, D. Tan, W. H. Brune, L. Jaeglé, D. J. Jacob, Y. Kondo, M. Koike, R. Chatfield, R. Pueschel, G. Ferry, G. Sachse, S. Vay, B. Anderson, J. Hannon and H. Fuelberg, J. Geophys. Res, 2000, 105(D3), 3771 CrossRef CAS.
- L. Jaeglé, D. J. Jacob, W. H. Brune, I. Faloona, D. Tan, B. G. Heikes, Y. Kondo, G. W. Sachse, B. Anderson, G. L. Gregory, H. B. Singh, R. Pueschel, G. Ferry, D. R. Blake and R. E. Shetter, J. Geophys. Res, 2000, 105(D3), 3877 CrossRef CAS.
- C. A. Cantrell, L. Mauldin, M. Zondlo, F. Eisele, E. Kosciuch, R. Shetter, B. Lefer, S. Hall, T. Campos, B. Ridley, J. Walega, A. Fried, B. Wert, F. Flocke, A. Weinheimer, J. Hannigan, M. Coffey, E. Atlas, S. Stephens, B. Heikes, J. Snow, D. Blake, N. Blake, A. Katzenstein, J. Lopez, E. V. Browell, R. Cohen, J. Thornton, R. Rosen, P. Wooldridge, D. Day, J. Dibb, E. Scheuer, G. Seid and R. Talbot, J. Geophys. Res., in the press Search PubMed.
- C. A. Cantrell, D. H. Stedman and G. J. Wendel, Anal. Chem., 1984, 56, 1496 CrossRef CAS.
- C. A. Cantrell, R. E. Shetter, J. A. Lind, A. H. McDaniel, J. G. Calvert, D. D. Parrish, F. C. Fehsenfeld, M. Buhr and M. Trainer, J. Geophys.Res., 1993, 98, 2897 CAS.
- P. S. Monks, L. J. Carpenter, S. A. Penkett and G. P. Ayers, Geophys. Res. Lett., 1996, 23, 535 CrossRef CAS.
- L. J. Carpenter, P. S. Monks, B. J. Bandy and S.A. Penkett, J. Geophys. Res., 1997, 102(D21), 25417 CrossRef CAS.
- S. A. Penkett, P. S. Monks, L. J. Carpenter, K. C. Clemitshaw, G. P. Ayers, R. W. Gillett, I. E. Galbally and C. P. Meyer, J. Geophys. Res., 1997, 102(D11), 12805 CrossRef CAS.
- P. S. Monks, L. J. Carpenter, S. A. Penkett, G. P. Ayers, R. W. Gillett, I. E. Galbally and C. P. Meyer, Atmos. Environ., 1998, 32, 3647 CrossRef CAS.
- P. S. Monks, G. Salisbury, G. Holland, S. A. Penkett and G. P. Ayers, Atmos. Environ., 2000, 34, 2547 CrossRef CAS.
- G. Salisbury, A. R. Rickard, P. S. Monks, B. J. Allan, S. Bauguitte, S. A. Penkett, N. Carslaw, A. C. Lewis, D. J. Creasey, D. E. Heard, P. J. Jacobs and J. D. Lee, J. Geophys. Res., 2001, 106(D12), 12669 Search PubMed.
- P. Zanis, P. S. Monks, E. Schuepbach and S. A. Penkett, J. Geophys. Res., 1999, 104(D21), 26913 CrossRef CAS.
- M. E. Jenkin, S. F. M. Ashbourn and K. C. Clemitshaw, Laboratory and Modelling Studies of the Peroxy Radical Chemical Amplification (PERCA) Technique, AEA Technology Report AEAT-4861/20147001/003, AEA Technology, Culham, Oxfordshire, UK, 1999 Search PubMed.
- T. J. Kelly, C. W. Spicer and G. F. Ward, Atmos. Environ., 1990, 24A(9), 2397 CAS.
- A. R. Curtis and W. P. Sweetenham, FACSIMILE Release H User’s Manual, UK Energy Authority, HARWELL, Computer Science and Systems Division, Harwell Laboratory, Oxfordshire, 1985 Search PubMed.
- C. E. Reeves, S. A. Penkett, S. Bauguitte, K. S. Law, M. J. Evans, B. J. Bandy, P. S. Monks, G. D. Edwards, H. Barjat, J. Kent, K. Dewey, S. Schmitgen and D. Kley, J. Geophys. Res, in the press Search PubMed.
- J. P. Abram, D. J. Creasey, D. E. Heard, J. D. Lee and M. J. Pilling, Geophys. Res. Lett., 2000, 27, 3437 CrossRef CAS.
- G. D. Edwards, PhD thesis, University of Leicester, UK, 2000.
- N. Brough, NOx and NOy measurements in Maximum Oxidation Rates in the Free Troposphere (MAXOX), Final Report ed. Ø. Hov, ENV4-CT97-0525, 2000pp 21–23 Search PubMed.
- C. M. Mihele and D. R. Hastie, Geophys. Res. Lett., 1998, 25(11), 1911 CrossRef CAS.
- C. M. Mihele, M. Mozurkewich and D. R. Hastie, Int. J. Chem. Kinet., 1999, 31, 145 CrossRef CAS.
- J. Burkert, M. D. A. Hernandez, D. Stöbener and J. P. Burrows, J. Geophys. Res, 2001, 106(D6), 5457 Search PubMed.
- W. B. DeMore, C. J. Howard, S. P. Sander, A. R. Ravishankara, D. M. Golden, C. E. Kolb, R. F. Hampson, M. J. Molina and M. J. Kurylo, Chemical Kinetics and Photochemical Data for Use in Stratospheric Modeling, Evaluation Number 12, JPL Publication 97-4, California Institute of Technology, Pasadena, California, 1997 Search PubMed.
- P. Zanis, P. S. Monks, E. Schuepbach, L. J. Carpenter, T. J. Green, G. P. Mills, S. Bauguitte and S. A. Penkett, J. Geophys. Res., 2000, 105(D19), 24223 CrossRef CAS.
- L. J. Carpenter, T. J. Green, G. P. Mills, S. Bauguitte, S. A. Penkett, P. Zanis, E. Schuepbach, N. Schmidbauer, P. S. Monks and C. Zellweger, J. Geophys. Res., 2000, 105(D11), 14547 CrossRef.
| This journal is © The Royal Society of Chemistry 2003 |