Direct measurements of urban OH reactivity during Nashville SOS in summer 1999
T. A.
Kovacs
a,
W. H.
Brune
a,
H.
Harder
a,
M.
Martinez
a,
J. B.
Simpas
a,
G. J.
Frost
b,
E.
Williams
b,
T.
Jobson
d,
C.
Stroud
c,
V.
Young
d,
A.
Fried
e and
B.
Wert
e
aDepartment of Meteorology, The Pennsylvania State University, University Park, PA, USA
bAeronomy Laboratory, National Oceanic and Atmospheric Administration and the Cooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, CO, USA
cYork University, North York, Ontario, Canada
dDepartment of Chemical Engineering, Ohio University, OH, USA
eAtmospheric Chemistry Division, National Center for Atmospheric Research, Boulder, CO, USA
First published on 8th August 2002
Abstract
Emissions of volatile chemicals control the hydroxyl radical (OH), the atmosphere's main cleansing agent, and thus the production of secondary pollutants. Accounting for all of these chemicals can be difficult, especially in environments with mixed urban and forest emissions. The first direct measurements of the atmospheric OH reactivity, the inverse of the OH lifetime, were made as part of the Southern Oxidant Study (SOS) at Cornelia Fort Airpark in Nashville, TN in summer 1999. Measured OH reactivity was typically 11 s−1. Measured OH reactivity was 1.4 times larger than OH reactivity calculated from the sum of the products of measured chemical concentrations and their OH reaction rate coefficients. This difference is statistically significant at the 1σ uncertainty level of both the measurements and the calculations but not the 2σ uncertainty level. Measured OH reactivity was 1.3 times larger than the OH reactivity from a model that uses measured ambient concentrations of volatile organic compounds (VOCs), NO, NO2, SO2, and CO. However, it was within ∼10% of the OH reactivity from a model that includes hydrocarbon measurements made in a Nashville tunnel and scaled to the ambient CO at Cornelia Fort Airpark. These comparisons indicate that 30% of the OH reactivity in Nashville may come from short-lived highly reactive VOCs that are not usually measured in field intensive studies or by US EPA's Photochemical Assessment Monitoring Stations.
Aim of investigation
The hydroxyl radical, OH, is the atmosphere's primary oxidant of chemical emissions from Earth's surface. While its production rate from solar photodestruction of ozone and other chemicals is large, so is its loss rate. In urban environments, typical OH lifetimes are 0.03–0.2 s. OH is lost by reactions with chemicals emitted from the surface and with chemical products of these oxidation reaction sequences. Thus the OH lifetime is dictated by the abundances of the chemical emissions and their oxidation products.The OH reactivity is the inverse of the OH lifetime. It is given by the equation:
| kOH = Σ kOH+VOCi [VOCi] + kOH+CO [CO] + kOH+NO [NO] + kOH+NO2 [NO2] + kOH+SO2 [SO2] + … | (1) |
Reactions between OH and the emitted chemicals initiate reaction sequences in which both ozone and secondary aerosols are produced. Reactions of HO2 and RO2 with NO produce NO2, which is broken apart by sunlight, leading to ozone formation. The instantaneous ozone production rate can be determined with measurements of HO2, RO2, and NO:
| P(O3) = {kHO2+NO [HO2] + ΣikRO2I+NO [RO2i]} [NO] | (2) |
Because HO2 and RO2 are not routinely measured, a proxy method using more routine measurements would be useful for routinely determining P(O3). In one proposed proxy method,1,2 the instantaneous O3 production rate can be calculated from the modeled primary HOx (HOx = OH + HO2) production, the OH reactivity that is calculated from measured chemicals and eqn. (1), the modeled yield of HO2 + RO2 per OH reaction, and the NOx (NOx = NO + NO2) abundance. This method applies for conditions in which NO is greater than a ppbv (ppbv = 10−9 by volume). The measurements required for this approach are available at the EPA Photochemical Assessment Monitoring Stations (PAMS), with the assumption that HOx production and OH reactivity can be correctly calculated.
The understanding of tropospheric oxidation chemistry is tested by intensive field campaigns. Instruments to measure meteorological variables, aerosols, and gas-phase chemicals are co-located for a month to six weeks. Simultaneous high-quality measurements from all instruments are usually obtained for only part of the intensive campaign period. These simultaneous measurements are then combined. Observed abundances of the gas-phase chemicals are then used to constrain point model calculations. The observed and calculated abundances of OH, HO2, and RO2 are then compared.
For several field campaigns, the observed OH was less than the modeled OH. Some investigators have presented the hypothesis that observed OH was lower than modeled because it was reacting with additional, unmeasured chemicals, probably volatile organic compounds (VOCs).3–7 While this hypothesis for the lower-than-expected OH is not the only one, it requires scrutiny because, if true, the current understanding of chemical emissions and their oxidation chemistry would need some modification. Recent chromatography measurements using a new technique suggest that traditionally unmeasured VOCs, particularly those with more than 6 carbon atoms, are contributing significantly to the OH reactivity in urban air.8
Two methods are typically used to determine the OH reactivity. The most common approach is to measure as many chemicals that react with OH as possible, multiply the concentration of each chemical by its reaction rate coefficient with OH, and then sum the products, as in eqn. (1). For this method to be valid, arguments must be made that the unmeasured chemicals – often difficult-to-measure large or oxygenated or highly reactive VOCs – do not contribute significantly to the sum. A second method is to calculate the OH loss rate using a photochemical model constrained by chemical measurements. However, since photochemical point models often contain lumped hydrocarbon oxidation mechanisms, accounting for all OH reactions can be difficult. Just as in the first method, models are limited by the VOC measurements that are put into the models. Until recently these were the only methods for determining OH reactivity.
A third method, one that enables tests of the hypothesis that unmeasured chemicals are reacting with OH, is now available. The OH reactivity can now be measured directly using techniques perfected for laboratory kinetics measurements over the last three decades.9,10 Direct measurements of OH reactivity were made for the first time during the Nashville SOS field intensive in summer 1999. In this manuscript, these direct OH reactivity measurements are compared to the calculated and the modeled OH reactivity to examine the issue of unmeasured and poorly modeled OH reactants.
Description of the experimental process
A new instrument, the total OH loss-rate measurement (TOHLM), directly measures the OH reactivity of air.9 Measurements of the OH reactivity are obtained with a technique similar to the laboratory kinetics discharge-flow technique.11,12 A large abundance of OH is introduced through a moveable injector into a flow tube in which a flow of ambient air has been established at a known velocity. This mixture is sampled downstream of the injector with a detection axis similar to those used to detect atmospheric OH. By changing the distance between the OH detector and the OH injection point, the OH reactivity (s−1) is measured.More specifically, air is pulled into a 5 cm diameter flow tube with an air speed of 0.4–0.8 m s−1, as measured by a hot wire anemometer (Fig. 1). OH and HO2 are created in a 1 cm diameter stainless steel tube by photolysis of water vapor in nitrogen (99.999%, <0.5 ppm THC), using the 185 nm radiation from a mercury lamp that is shielded from the airflow. The OH and nitrogen are then injected into the flow tube through holes that were drilled radially in a Teflon endcap. Typical OH mixing ratios are 10–100 pptv (pptv = 10−12 by volume), high enough to avoid influences from ambient HOx levels, where HOx = OH + HO2, and low enough to avoid complications from HOx self-reactions and the depletion of OH reactants.9 The injected OH mixes rapidly across the flow tube and reacts with the chemicals in the ambient air that was pulled into the flow tube. At the flow tube's end, OH is detected by laser-induced fluorescence in a low-pressure detection chamber.13,14 After a total time of 20 s (10 s measuring OH + laser-generated background signals and 10 s measuring laser-generated background signals), the injector is pulled back. The reaction time is increased because the mixture now has to travel farther down the tube at a constant velocity before it is sampled. Measurements are made in 10 steps of 1–2 cm each, giving a total reaction time of 0.15–0.4 s. The time to measure each decay is approximately 4 min. For typical conditions, the OH signal decays a factor of 10–20 in this reaction time.
 | ||
| Fig. 1 Schematic of the total OH loss-rate measurement instrument (TOHLM). The detection system consists of a laser beam crossed with a detection axis that includes a microchannel plate detector (MCP), which detects the OH fluorescence in the lower pressure detection chamber. The hot wire anemometer monitors the air speed in the flow tube. The zigzags indicate that the flow tube is longer relative to the diameter. | ||
The OH reactivity, kOH, is ideally determined by the slope of the logarithm of the OH signal, SOH, as a function of time minus the first order rate of OH loss to the flow tube's walls:
| kOH = − Δ ln(SOH)/Δ time − kwall | (3) |
The OH mixing was tested by adding CO through the injector into zero air and measuring the uniformity of the CO mixing ratio across the flow tube. Its uniformity was better than 10%. The velocity measurements with a hot wire anemometer were verified by comparing the velocity measurement integrated across the flow tube to mass flow, as measured by the pressure change in a zero-air cylinder in a fixed amount of time. The two methods agreed to within 10%. The velocity profile across the flow tube was measured with a hot wire anemometer to be flat to within ±10%. Radial and axial diffusion, which could affect the OH reactivity measurement, are calculated to be insignificant for these atmospheric pressure flows.9
The loss of OH on the flow tube walls must be subtracted from the observed decays in order to calculate the OH reactivity. To test for wall loss, zero air (hydrocarbons < 0.5 ppmv, from several vendors) was introduced into TOHLM in the place of ambient air. For the 5 cm diameter flow tube used in Nashville, the OH loss on the bare glass wall, kwall, was 5.1 ± 0.6 s−1 (1σ confidence). Although the wall loss was measured to be the same in zero air of different quality from different vendors, it is possible that some of the OH loss was due to impurities in the zero air and that the wall loss is actually less. This OH wall loss could have been reduced using wall treatments such as Teflon, but then, the wall loss may have varied depending on the levels of ambient pollutants that might coat the walls. This variable wall loss is much less desirable than a larger but steady wall loss. Wall loss tests that were made throughout the Nashville study demonstrated that the OH wall loss did not change and thus was simple to take into account in the analyses of the OH signal decays.
While the mixing, velocity, velocity profile, and OH wall loss were carefully studied to reduce uncertainties, the proof of TOHLM's ability to accurately measure OH reactivity comes from the comparison of the measured OH reactivity with the calculated OH reactivity for known amounts of CO or hydrocarbons that were added either to the ambient flow or to zero air.9 Typically, these values agree to better than 15%, with an average difference of less than 10%. By calibrating with different gases that have widely differing rate coefficients, we have determined that the absolute uncertainty of TOHLM is ±14% (1σ confidence). The total uncertainty in the OH reactivity measurements comes from a propagation of error analysis that combines the absolute uncertainty with the uncertainty in the OH wall loss. The total uncertainty is ±1.5 s−1 (1σ confidence) when the OH reactivity is 10 s−1.
An important consideration for any OH reactivity measurement is the recycling of the HO2, formed in the reaction of OH with CO and VOCs, by the reaction:
| HO2 + NO → OH + NO2 | (4) |
| d[OH]/dt = − kOH [OH] − kwall [OH] + kNO+HO2 [NO] [HO2] | (5) |
| kOH = − Δ ln(SOH)/Δ time − kwall + kNO+HO2 [NO] SHO2/SOH | (6) |
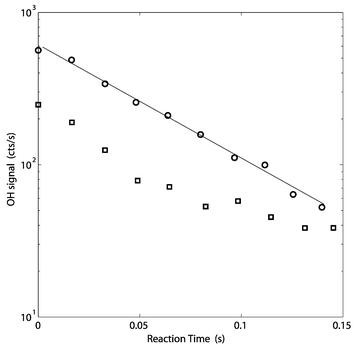 | ||
| Fig. 2 Typical OH decays from 6 July 1999. The lower decay (squares) was at 8∶35 CST (Central Standard Time, GMT − 6 h), when NO was ∼20 ppbv; the upper linear decay was at 9∶20 CST when NO was ∼1 ppbv. The upper decay is linear for more than a factor of 10; the lower decay shows clearly the curvature that results from HO2 + NO → OH + NO2. | ||
HO2 signal decays were measured at least twice each day in Nashville. While photolysis of water produces OH and HO2 in equal amounts inside the injector, the ratio of HO2 to OH ratio at the measurement distance closest to the detector was 3.0 ± 1.6 and increased at greater distances. This difference is a result of more OH than HO2 being lost inside the injector and the endcap prior to being injected into the flow tube. Because all the factors in the last term in eqn. (6) were measured, the decays of SOH can be corrected for the HO2 recycling prior to the application of eqn. (3) to find kOH in the OH reactivity retrieval algorithm. The resulting corrected kOH was less than 1.05 times larger than the observed kOH when NO was <0.5 ppbv, 1.08 times larger when NO was 1 ppbv, 1.4 times larger when NO was 5 ppbv, and 2 times larger when NO was 10 ppbv.
OH decays generated by a simple kinetic model were used to test the algorithm's ability to accurately retrieve kOH. The values for the OH and HO2 signals, reaction time steps, kOH, and NO were all chosen to match typical values encountered during the Nashville study. Model kOH ranged from 5 to 15 s−1 and NO from 0 to 20 ppbv. The retrieval algorithm produced kOH values that agreed to within 7% of the model kOH values over the entire range of conditions. For NO less than 5 ppbv, the agreement was better than 3%. Thus, accurate OH reactivity values can be retrieved even when NO is 20 ppbv, which occurs often in urban areas.
For this retrieval to be accurate, the NO mixing ratio and the ratio SHO2/SOH must be accurately known. If initial HO2/OH signal ratios of 2.0 and 4.5 are used in the simple kinetic model to generate decay curves, instead of 3.0, the retrieved values for kOH increasingly deviate from the initial model kOH values as NO is increased. At NO = 5 ppbv, the deviation is 15–20%. This deviation is a measure of the retrieval uncertainty caused by the uncertainty in SHO2/SOH. This uncertainty could have been greatly reduced had HO2 decays been measured more often in Nashville. When this uncertainty is combined with the uncertainties for the measured NO and kNO+HO2, the total additional uncertainty in the measured OH reactivity is 0.85 s−1 (1σ confidence) when kOH = 10 s−1 and NO = 5 ppbv and is 2.5 s−1 when NO = 10 ppbv. Thus for the Nashville study, OH reactivity is reported only when NO < 5 ppbv, conditions for which we deem the uncertainty to be acceptably small.
Wind and shifts in air mass chemical composition sometimes occurred during the ∼4 min that were required for each decay measurement. As a result, the affected decays look noisy. Such decays were rejected by a data filter that required that the fit to decay have an r2 of at least 0.8 when 7 points were included in the fit. Less than 5% of the decays were rejected by this criterion.
Observations at Cornelia Fort Airpark, Nashville
The measurements discussed in this paper took place at Cornelia Fort Airpark (CFA), about 8 km northeast of downtown Nashville, TN, during the Southern Oxidants Study (SOS 99) in June and July 1999. The site and its characteristics are described for a previous SOS study in 1995.15 Winds at CFA were generally from the south–southwest, although winds from the north–northwest occurred about a third as often. Surrounded by mixed deciduous forests and pastures, Nashville is a site particularly well suited to study interactions of anthropogenic and biogenic emissions.During the SOS campaign the total OH loss-rate measurement (TOHLM) instrument took data for much of 15 days from 26 June to 11 July. The OH signal was typically followed for more than an order of magnitude (Fig. 2).
Co-located at the Cornelia Fort Airpark were simultaneous measurements of meteorology, atmospheric chemical composition, and aerosols. Most important for analysis of OH reactivity were measurements of O3, NO, NO2, CO, SO2, which were made every minute for the entire period of OH reactivity measurements, and volatile organic compounds (VOCs), most of which were measured hourly. The Ohio University (OU) group measured hourly 24 VOCs from 5 July to 14 July, overlapping with the OH reactivity measurements for 7 days. The NOAA Aeronomy Laboratory (NOAA AL) and NCAR measured hourly a smaller set of VOCs, including oxidation products not otherwise measured, from 19 June to 28 June and again from 1 July to 14 July, overlapping with the OH reactivity measurements for 13 days. For this paper, the OU, NOAA AL, and NCAR measurements (Table 1) were used to calculate OH reactivity and as inputs to photochemical models.
| a (m) = 1 min. All others hourly. | ||
|---|---|---|
| Ethane | trans-2-Butene | Formaldehyde |
| Propane | Isoprene | Acetaldehyde |
| i-Pentane | cis-2-Pentene | Propanal |
| n-Pentane | trans-2-Pentene | i-Butanal |
| i-Butane | Benzene | n-Butanal |
| n-Butane | Toluene | Ethanol |
| 2-Methyl pentane | Ethyl benzene | Isopropanol |
| 3-Methyl pentane | o-Xylene | Carbon monoxide (m) |
| Acetylene | 1,3-Butadiene | Ozone (m) |
| Ethene | m,p-Xylene | Sulfur dioxide (m) |
| Propene | Acetone | Nitrogen dioxide (m) |
| 1-Butene | Methylvinylketone | Nitric oxide (m) |
| cis-2-Butene | Methylethylketone | Hydrogen peroxide (m) |
| Methylperoxide (m) | ||
The uncertainty in the calculated OH reactivity must take into account the uncertainties in the measured chemical concentrations and in the reaction rate coefficients. Measurements of the most important contributors to the OH reactivity have the following concentration uncertainties: NO, ±19%; NO2, ±22%;16 HCHO, ±10%;17 CO, ±15%; and VOCs, ±20%. CH4 was assumed to be 1750 ppbv. The typical uncertainty for the reaction rate coefficients with these species is 15% to 30%. Taken in quadrature, the uncertainty in the sum of the products of individual reactants and their reaction rate coefficients with OH is approximately ±1.8 s−1 (1σ confidence).
Results
Directly measured OH reactivity shows a small diurnal variation when all observations are averaged over a diurnal cycle (Fig. 3). The median and standard deviation values are (11.3 ± 4.8) s−1 for all measurements, (10.2 ± 2.5) s−1 for midday (10–16 CST), (13.6 ± 5.2) s−1 for morning rush hour (6–10 CST), and (11.6 ± 5.2) s−1 for night (20–4 CST). Mean values are less than 10% larger than median values. Restricting the OH reactivity values to times when NO < 5 ppbv does not seriously bias the results. NO was greater than 5 ppbv only 12% of the time and only 25% of the time during the morning rush hour. The diurnal curve for OH reactivity shows the expected peaks for the morning and evening rush hours, when chemical emissions are high and the planetary boundary layer is not at its maximum midday height. | ||
| Fig. 3 Measured OH reactivity (s−1) in Nashville SOS 1999. Hourly median values (black line) are calculated for the entire SOS study. Individual points (gray) show the scatter from day to day. Error bars are ±1.5 s−1 uncertainty (1σ confidence). | ||
The midday OH reactivity varied little from day to day, as can be seen from the small standard deviation. On the other hand, the nocturnal OH reactivity showed large day-to-day variability. The nocturnal variability is driven by a few nights in which the OH reactivity exceeded 15 s−1. The higher values for nocturnal OH reactivity occurred for all nocturnal wind directions, which were only from the north, west, and south. Thus, the large nocturnal OH reactivity does not appear to have come from a single large source. It tended to occur when wind speeds are less than 4 m s−1. Variations in the stability of the nocturnal boundary layer are a likely cause of the observed variability in the nocturnal OH reactivity.
Part of the increased nocturnal OH reactivity came from the variable nocturnal NO2, which contributed more than 2 s−1 to the OH reactivity for 60% of the nocturnal measurements. During the night, the reaction between OH and NO2 was 15% to 40% of the total calculated OH reactivity, while during the daytime, it was less than 10%. Even when the OH reactivity due to NO2 is subtracted, the highest nocturnal OH reactivity values still occur for all nocturnal wind directions and for wind speed is < 4 m s−1.
Comparison to calculated OH reactivity
The calculated OH reactivity is determined by eqn. (1), using measured chemical concentrations (Table 1) and reaction rate coefficients.18–23 The combined OU, NOAA AL, and NCAR measurements overlap with measured OH reactivity for only 9 points in July. If only the VOCs that contribute significantly to the calculated OH reactivity are considered, the number of overlap points increases to 240. These most significant VOCs were isoprene, acetaldehyde, propanal, i-butanal, n-butanal, ethene, n-pentane, methacrolein, methyl vinyl ketone, and ethanol. The mean contribution of each other individual VOC was less than 0.1% of the mean calculated total OH reactivity; when summed together, these less significant VOCs contributed ∼3%. To account for this OH reactivity, an additional 3% contribution was added to all the calculated OH reactivities determined from only the most significant contributors, with the assumption that the contributions of the less-significant VOCs were not more important at the times when they were not measured.The median OH reactivity and standard deviations calculated from measured chemicals from OU and NOAA AL was (7.2 ± 3.5) s−1 for all measurements, (5.6 ± 1.2) s−1 during midday and (8.5 ± 4.3) s−1 at night (Fig. 4). The major calculated OH reactivity during the day was by reactions with CO (∼1 s−1), isoprene (∼1 s−1), formaldehyde (∼1 s−1), other carbonyls (∼1 s−1), NO2 (∼1 s−1), VOCs (∼2 s−1), and CH4 (0.3 s−1). NO2 had a larger calculated contribution at night when NO2 abundances climbed to 20–50 ppbv, contributing 5–13 s−1 to the calculated OH reactivity.
 | ||
| Fig. 4 Comparison of median measured OH reactivity (s−1) (dashed line, circles) to median calculated OH reactivity (solid line) for the entire SOS study. The 240 individual calculated points are in gray. Measured OH reactivity is selected for coincidence with calculated OH reactivity. 1σ error bars are ±1.8 s−1 for the calculated OH reactivity (solid lines) and ±1.5 s−1 for the measured OH reactivity (dashed lines). | ||
A comparison of the measured and calculated OH reactivity shows that the measured OH reactivity was greater than the calculated OH reactivity by a mean difference of (3.8 ± 2.0) s−1. The mean ratio of calculated-to-measured OH reactivity is 0.69 ± 0.12. This difference is statistically significant (2σ confidence) by a statistical t-test even if we assume that the TOHLM calibration is actually lower by the 1σ uncertainty (1.5 s−1) and the calculated OH reactivity is actually higher by the 1σ uncertainty (1.8 s−1). It is not statistically significant if we assume that the TOHLM calibration is actually lower by the 2σ uncertainty and the calculated OH reactivity is actually higher by the 2σ uncertainty. In Fig. 4, the median measured OH reactivity has been determined for only those measurements that coincide with calculated OH reactivity. Thus, it is appears that, in Nashville, the calculated OH reactivity may fail to account for about 30% of the actual OH reactivity as determined by TOHLM.
Comparison to modeled OH reactivity
Models with explicit reactions can be used to determine modeled OH reactivity. Modeled OH reactivity should be greater than the calculated OH reactivity because the model will account for reactions between OH and reactive chemicals that are difficult to measure. However, the modeled OH reactivity should not be significantly greater than the calculated OH reactivity, since most short-lived VOC oxidation products should react with O2 or NO before they can with the less abundant OH. The Regional Atmospheric Chemistry Mechanism (RACM)24 was used in the FACSIMILE photochemical modeling program to calculate the OH reactivity.25 The model was constrained by measurements of all observed chemicals including the VOCs used in the calculated OH reactivity, but not the measurements of OH and HO2.Modeled and measured OH reactivity have only 25 overlapping data points. The most significant result is that the modeled OH reactivity is 9% larger than the calculated OH reactivity. As a result, the model suggests that reactive short-lived VOCs account for less than 1 s−1 of additional OH reactivity. The modeled OH reactivity is still more than 20% lower than the measured OH reactivity.
Comparison to tunnel VOC measurements
Another way to examine the potential contribution of short-lived VOCs to the OH reactivity is to use the VOC measurements in a Nashville tunnel.26 In the roadway tunnel, the levels of all vehicle emissions, including several short-lived VOCs, are increased above instrument detection limits and can thus be quantified, whereas they are not detectable in open air. These short-lived VOCs include long straight and branched chain alkanes and internal and external alkenes. The abundances of these short-lived VOCs at CFA can be estimated by scaling them to the CO measured in the tunnel and then using the measured CO at CFA to derive short-lived VOC values for CFA. Scaling to CO makes sense because vehicles are thought to account for ∼90% of CO in urban areas.27The tunnel data were incorporated into the NOAA AL model, which uses a chemical mechanism with lumped groups of chemical species, similar to RACM. The model incorporated measurements made at CFA, including the mean measurements of isoprene, α-pinene, and higher carbonyls, and the measurements of NO, NO2, CO, HCHO, and all other chemical species measured with a time resolution of a few minutes or less. The light, unreactive alkanes were derived directly from the CFA measurements using correlations of the hourly CFA data with the measured CO. The modeled OH reactivity using just these chemicals is similar to the RACM modeled OH reactivity. The main difference between this calculation of OH reactivity and the previous ones is the inclusion of short-lived VOCs at abundances relative to CO that occur just after being emitted from vehicles.
The reactive VOCs emitted by vehicles have short lifetimes, typically 20 minutes to a few hours, and thus will decrease more rapidly with distance from the sources than will CO.28 Because the composition of the urban air should be a mixture of chemicals emitted at different distances from Cornelia Fort Airpark, the scaling to CO from the tunnel data should give an upper limit to the OH reactivity from vehicles, if all the important chemical species were measured in the tunnel. On the other hand, vehicle emissions are only part of Nashville's VOC mix; stationary and biogenic sources are also present. This procedure does not account for unmeasured, reactive VOCs from stationary and biogenic sources, although these are thought to make only small contributions. Despite these caveats, agreement between the measured OH reactivity and that derived from tunnel data would indicate the potential importance of these short-lived VOCs to the actual OH reactivity at Cornelia Fort Airpark.
When these short-lived chemicals, with abundances scaled to CO, are added to the calculation of OH reactivity, the median tunnel calculated OH reactivity (hereafter called tunnel OH reactivity) and its standard deviation is (9.0 ± 4.3) s−1 for all measurements, (8.6 ± 1.7) s−1 during midday and (11.1 ± 5.0) s−1 at night (Fig. 5). A comparison of the median measured and tunnel OH reactivity shows that the tunnel OH reactivity is on average 0.87 ± 3.6 s−1 below the measured OH reactivity; the mean ratio of tunnel-to-measured OH reactivity is 0.94 ± 0.18. In Fig. 5, the median measured OH reactivity has been determined for only those measurements that coincide with calculated OH reactivity. Even the median diurnal variation is the same for the measured and tunnel OH reactivity. Thus in Nashville, the tunnel OH reactivity agrees much better with the measured OH reactivity than either the calculated or the modeled OH reactivity.
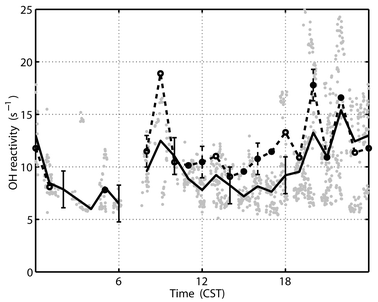 | ||
| Fig. 5 Comparison of median measured OH reactivity (s−1) (dashed line, circles) to median tunnel OH reactivity (solid line) for the entire SOS study. The 546 individual tunnel calculated OH reactivity points are in gray. Measured OH reactivity is selected for coincidence with tunnel OH reactivity. 1σ error bars are ±1.8 s−1 for the tunnel OH reactivity (solid lines) and are ±1.5 s−1 for the measured OH reactivity (dashed lines). | ||
Discussion
TOHLM provides an excellent check on the possibility that unmeasured, short-lived VOCs are significant contributors to urban OH reactivity.8 For Nashville in summer 1999, the measured OH reactivity is greater than that calculated using the measurements of chemicals that react with OH, although this difference is statistically significant at the 1σ, but not the 2σ, uncertainty level of both the measurements and the calculations. Modeled OH reactivity is also less than the measured OH reactivity. Only when short-lived VOCs, measurable only in a Nashville tunnel, are included in the calculation does the calculated OH reactivity agree to within a 10% of measured OH reactivity. The overall conclusion is that the typical method of calculating OH reactivity from the measured VOCs probably underestimated the actual OH reactivity, which was determined by TOHLM, for Nashville in summer 1999.What are the effects of this possible underestimation of the OH reactivity in Nashville in summer 1999? First of all, estimates of ozone production will be 30% low if they are based on the calculated OH reactivity with VOCs.1 Second, increased VOC reactivity will increase the OH chain length for given values of NOx and O3, where chain length is defined as {kOH+CO[CO] + ΣkVOCi+OH [VOCi]}/kNO2+OH [NO2]. The OH reaction with CO and VOCs cycles OH while the OH reaction with NO2 forms HNO3 and terminates OH. Third, as a result of increased chain length, the peak ozone production will be shifted to NOx values that are 1.4 times higher than currently assumed, provided that the peroxide formation rates are correct and only NO and O3 cycle HO2 back to OH. Finally, under conditions where HOx cycling between OH and HO2 is faster than HOx production, the measured HO2/OH ratio should be 1.3 times larger than modeled, since [HO2]/[OH] = {kOH − kNO2+OH[NO2]}/{kNO+HO2[NO] + kO3+HO2[O3]} and measured kOH is 1.3 times larger than modeled. The under-representation of the OH reactivity may have other impacts.
The difference between measured and calculated OH reactivity in Nashville SOS is significant at only the 1σ uncertainty level because of the measured, calculated, and modeled uncertainties. However, it is possible that unmeasured reactive VOCs may contribute more significantly to the OH reactivity in other environments or conditions. Since Nashville SOS 1999, TOHLM measurements have been made as part of field intensive campaigns in a Michigan rural forested environment and two large urban areas, Houston, Texas, and Queens, New York. The results of these intensive field studies are now being analyzed. If the differences between measured and calculated OH reactivity vary from environment to environment, a likely culprit for the differences would be unmeasured, short-lived VOCs that also vary from environment to environment.
The OH reactivity that was measured by the Nashville version of the TOHLM had a lower uncertainty than either the calculated or the modeled OH reactivities. The current version of TOHLM has an even lower 1σ uncertainty, approximately ±1.3 s−1 for a OH reactivity of 10 s−1, and further improvements will likely reduce the uncertainty to ±1 s−1 or less. It is unlikely that the calculated or modeled OH reactivities will ever have 1σ uncertainties that low. In addition, for some environments, the chemicals that contribute significantly to OH reactivity may be either unmeasured or poorly measured. Because TOHLM is a more accurate measure of OH reactivity than either calculated or modeled OH reactivity, its widespread deployment would improve the understanding of tropospheric photochemistry and would provide additional scientific rationale for regulatory actions to mitigate urban and regional ozone pollution.
Acknowledgements
We thank NSF grant ATM-9974335 and NOAA grant 40RANR901074 for support for this work. Robert Lesher, Jeremy Bassis, and Xinrong Ren were crucial to the success of these measurements and the analyses of the data. The comments of the two anonymous reviewers have resulted in a better analysis and manuscript. We thank J. Meager and F. Fehsenfeld for inviting us to participate in Nashville SOS 1999. We also thank all the other research groups that made Nashville SOS 1999 a success.References
- L. I. Kleinman, P. H. Daum, J. H. Lee, Y-N. Lee, L. J. Nunnermacher, S. R. Springston, L. Newman, J. Weisenstein-Lloyd and S. Silman, Geophys. Res. Lett., 1997, 24, 2299–2302 CrossRef CAS.
- P. H. Daum, L. I. Kleinman, D. Imre, L. J. Nunnermacher, Y.-N. Lee, S. R. Sprinston and L. Newman, J. Geophys. Res., 2000, 105, 9107–9119 CrossRef CAS.
- D. Poppe, J. Zimmermann, R. Bauer, T. Brauers, D. Bruening, J. Callies, H.-P. Dorn, A. Hofzumahaus, F. J. Johnen, A. Khedim, H. Koch, R. Koppermann, H. London, K.-P. Mueller, R. Neuroth, C. Plass-Duelmer, U. Platt, F. Rohrer, E.-P. Roeth, J. Rudolph, U. Schmidt, M. Wallasch and D. H. Ehhalt, J. Geophys. Res., 1994, 99, 16,633–16,642 CrossRef CAS.
- F. Eisele, D. Tanner, C. Cantrell and J. Calvert, J. Geophys. Res., 1996, 101, 14,665 CrossRef CAS.
- S. McKeen, G. Mount, F. Eisele, E. Williams, J. Harder, P. Goldan, W. Kuster, S. Liu, K. Baumann, D. Tanner, A. Fried, S. Sewell, C. Cantrell and R. Shetter, J. Geophys. Res., 1997, 102, 6467 CrossRef CAS.
- P. Stevens, J. Mather, W. Brune, F. Eisele, D. Tanner, A. Jefferson, C. Cantrell, R. Shetter, S. Sewell, A. Fried, B. Henry, E. Williams, K. Baumann, P. Goldan and W. Kuster, J. Geophys. Res., 1997, 102, 6379 CrossRef CAS.
- R. L. Mauldin III, G. Frost, G. Chen, D. Tanner, A. Prevot, D. Davis and F. Eisele, J. Geophys. Res., 1998, 103, 16,713.
- A. C. Lewis, N. Carslaw, P. J. Marriot, R. M. Kinghorn, P. Morrison, A. L. Lee, K. D. Bartle and M. J. Pilling, Nature, 2000, 405, 778–781 CrossRef CAS.
- T. A. Kovacs and W. H. Brune, J. Atmos. Chem., 2001, 39, 105–122 CrossRef CAS.
- F. Jeanneret, F. Kirchner, A. Clappier, H. van den Bergh and B. Calpini, J. Geophys. Res., 2001, 106, 3083–3093 Search PubMed.
- F. Kaufman, J. Phys. Chem., 1984, 88, 4909 CrossRef CAS.
- N. Donahue, J. Clarke, K. Demerjian and J. Anderson, J. Phys. Chem., 1996, 100, 5821 CrossRef CAS.
- J. H. Mather, P. S. Stevens and W. H. Brune, J. Geophys. Res., 1997, 102, 6427–6436 CrossRef CAS.
- D. Tan, I. Faloona, J. B. Simpas, W. Brune, J. Olson, J. Crawford, M. Avery, G. Sachse, S. Vay, S. Sandholm, H.-W. Guan, T. Vaughn, J. Mastromarino, B. Heikes, J. Snow, J. Podolske and H. Singh, J. Geophys. Res., 2001, 106, 24,407–24,427 Search PubMed.
- E. B. Cowling, W. L. Chameides, C. S. Kiang, F. C. Fehsenfeld and J. F. Meager, J. Geophys. Res., 1998, 103, 22,209–22,212 CrossRef CAS.
- E. Williams, K. Baumann, J. Roberts, S. Bertman, R. Norton, F. Fehsenfeld, S. Springston, L. Nunnermacker, L. Newman, K. Olszyna, J. Meagher, B. Hartsell, E. Edgerton, J. Pearson and M. Rodgers, J. Geophys. Res., 1998, 103, 22,261 CrossRef CAS.
- B. Wert, A. Fried, B. Henry, J. Drummond, S. Rauenbuehler, J. Walega, W. Potter and J. Stutz, J. Geophys. Res., 2002 Search PubMed , in preparation.
- R. Atkinson, J. Phys. Chem. Ref. Data, Monograph, 1, 1989, 1–246 Search PubMed.
- R. Atkinson, J. Phys. Chem. Ref. Data, Monograph, 2, 1994, 1–216 Search PubMed.
- R. Atkinson, J. Phys. Chem. Ref. Data, 1997, 26, 215–290 CAS.
- R. Atkinson, D. Baulch and J. Troe, J. Phys. Chem. Ref. Data, 1999, 28, 191 CAS.
- W. B. DeMore, S. P. Sander, D. M. Golden, R. F. Hampson, M. J. Kurylo, C. J. Howard, A. R. Ravishankara, C. E. Kolb and M. J. Molina, Chemical kinetics and photochemical data for use in stratospheric modeling, NASA, Evaluation 12, JPL, Pasadena, CA, 1997 Search PubMed.
- S. P. Sander, R. R. Friedl, W. B. DeMore, D. M. Golden, R. F. Hampson, M. J. Kurylo, R. E. Huie, G. K. Moortgat, A. R. Ravishankara, C. E. Kolb and M. J. Molina, Chemical kinetics and photochemical data for use in stratospheric modeling, NASA, Supplement to Evaluation 12: Update of key reactions, JPL, Pasadena, CA, 2000 Search PubMed.
- W. R. Stockwell, F. Kirchner and M. Kuhn, J. Geophys. Res., 1997, 102, 25,847–25,879 CrossRef CAS.
- M. Martinez, H. Harder, T. Kovacs, J. B. Simpas, J. Bassis, W. H. Brune, E. J. Williams, C. A. Stroud, S. R. Hall, R. E. Shetter, B. Wert, A. Fried, B. Alicke and J. Stutz, J. Geophys. Res., 2002 Search PubMed , to be submitted.
- R. A. Harley, S. A. McKeen, J. Pearson, M. O. Rodgers and W. A. Lonneman, J. Geophys. Res., 2001, 106, 3559–3567 Search PubMed.
- Rethinking the Ozone Problem in Urban and Regional Air Pollution, Committee on Tropospheric Ozone Formation and Measurement, National Research Council, Washington, D.C., 1991 Search PubMed.
- D. D Parrish, M. Trainer, V. Young, P. D. Goldan, W. C. Kuster, B. T. Jobson, F. C. Fehsenfeld, W. A. Lonneman, R. D. Zika, C. T. Farmer, D. D. Riemer and M. O. Rodgers, J. Geophys. Res., 1998, 103, 22,339–22,359 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |