Recent developments in computational actinide chemistry
Nikolas
Kaltsoyannis
Department of Chemistry, University College London, 20 Gordon Street, London, UK WC1H 0AJ. E-mail: n.kaltsoyannis@ucl.ac.uk
First published on 31st October 2002
Abstract
This review describes recent computational investigations into the electronic and geometric structures of molecular actinide compounds. Following brief introductions to (i) the effects of relativity in chemistry and (ii) ab initio and density functional quantum chemical methods, four areas of contemporary research are discussed. These are π backbonding in uranium complexes, the geometric structures of bis benzene actinide compounds, the valence electronic structure of the uranyl ion, and the inverse trans influence in pseudo-octahedral [AnOX5]n−. Comparisons are made with experimental studies, and similarities and differences between d- and f-block chemistry are highlighted.
 Nikolas Kaltsoyannis Nikolas Kaltsoyannis | Nik Kaltsoyannis studied for his BA and DPhil degrees at Oxford University between 1985 and 1992. His doctoral supervisor was Professor Jennifer Green. He subsequently held a Postdoctoral Fellowship at the University of Ohio State with Professor Bruce Bursten before moving to the Lawrence Berkeley National Laboratory to take up a NATO Postdoctoral Fellowship with Dr Norman Edelstein. He joined the Department of Chemistry at University College London in 1994, where he is currently Senior Lecturer. The principal focus of his research is the computational study of the electronic and geometric structures and reactivity of molecular compounds of the d- and f-elements. |
1 Introduction
The actinide elements (elements 90–103) are the last complete family in the Periodic Table.1 They are all radioactive, and bar the first three (thorium, protactinium and uranium) are man-made. The use of uranium and plutonium in nuclear weapons and nuclear power generation lends these two elements a special chemical and physical significance. More specifically, the critical choices facing us over the correct way to dispose of uranium and plutonium (and their radioactive daughters) in the medium to long term means that we must understand as much as possible about the chemistry of these elements in particular, and the actinides in general.Given the constraints imposed on experimental chemistry by the toxicity, radioactivity and scarcity of the actinides, computational techniques are an increasingly important tool in their study. Quantum chemistry faces particular challenges when describing the actinides.2,3 Actinide complexes are frequently open-shell (due in part to there being many metal valence atomic orbitals (AOs) which lie close together in energy (5f, 6p, 6d, 7s)). The correct description of electron correlation effects (a key facet of reliable quantum chemistry) is extremely important (and difficult) in these cases. Furthermore, relativistic effects are large. Indeed, the incorporation of relativity into quantum chemical studies of actinide systems is essential in order to gain even a qualitative understanding of their electronic structure.
In this article I discuss four case studies, each describing recent computational work in a different area of molecular actinide chemistry. Wherever possible, comparisons are drawn with experimental research, and with the chemistry of related transition metal systems, with which the reader is probably more familiar. The aim of these latter comparisons is to illustrate some of the similarities and differences between the d- and the f-block, using important concepts in transition metal chemistry (e.g. the Dewar–Chatt–Duncanson model of synergic bonding and the trans influence) as a vehicle.
In order that the article be self-contained, I have included brief introductions to the effects of relativity in chemistry and to the computational techniques employed in the case studies. Readers familiar with these issues may skip the section entitled ‘Background material’ with no loss of continuity. For those readers who are unfamiliar with this material, I hope I have provided enough background to enable understanding of the case studies I have chosen. Further background information is contained in the textbooks and review articles referenced in this section.
2 Background material
2.1 Relativistic effects
The effects of relativity upon the physical and chemical properties of heavy elements are well established and documented.4,5 Relativistic effects may be divided into two categories; the modification of electronic wavefunctions and energies (i.e. of AOs), and spin-orbit coupling. The first effect can itself be split into two; direct orbital contraction and indirect orbital expansion. The former applies primarily to all s and, to a lesser extent, p orbitals, and is usually4–6 explained as follows. The inner core electrons move with radial velocities that are appreciable fractions of the speed of light. These high velocities lead to modifications in electron mass and radial extension, producing a contraction of the orbital. The AOs of the same l but higher n value then also contract to ensure orthogonality with the core functions.Unfortunately, this popular valence/core orthogonality argument is most likely incorrect. Baerends et al.7 have shown that the orthogonalisation of high n (valence) s and p functions on the core AOs actually leads to a small expansion of the valence orbitals. The overall contraction of these valence orbitals is in fact due to the mixing in of orbitals higher in energy (especially continuum orbitals) by the relativistically modified Hamiltonian.
The indirect orbital expansion describes the effect of relativity on valence d and f functions. It arises from increased shielding of the nucleus as a result of the direct contraction of the outer core s and p electrons of similar radial distribution to the d and f functions.
For very heavy elements such as the actinides, the relativistic modification of the valence AOs is very significant, and computational treatments of the 5f elements should really incorporate these AO modifications to the greatest extent feasible.
The relativistic analogue of the Schrödinger equation, formulated by Dirac, leads in a natural way to the concept of electron spin (which does not appear in non-relativistic quantum mechanics and must therefore be treated as an extra). Formally, all atomic and molecular wavefunctions are characterised by a total angular momentum which is the resultant of the electron’s intrinsic spin angular momentum and that imposed by its orbital motion. As these spin-orbit coupling effects have a very strong dependence on nuclear charge, they can be very significant for the actinides. For example, the interpretation and calculation of actinide optical spectra5,8 require that spin-orbit coupling be properly accounted for.
2.2 Computational techniques
Modern molecular quantum chemistry is largely the domain of two distinct theoretical approaches, the ab initio Hartree–Fock Self-Consistent-Field (HF-SCF) method (and its extensions) and density functional theory (DFT). This duality of approach extends to computational actinide chemistry, with the methods modified to take account of the effects of relativity. In this section I shall say a few words about both of these methods and make some general observations about their application to the 5f elements. For more information I refer the reader to references 6 and 9–11, whilst noting that there are many other excellent textbooks and review articles on computational quantum chemistry.The HF-SCF equations must be solved iteratively because the Hamiltonian operator of the HF-SCF method (the Fock operator) depends upon the basis functions on which it acts. Thus HF-SCF calculations begin by guessing a molecular electronic structure, and then apply the Fock operator to generate a new electronic structure. Once the electronic structure on two consecutive iterative cycles differs by less than a very small (and predefined) amount, the electronic structure is described as being self-consistent and the calculation terminates.
One of the key results of a HF-SCF calculation is the total molecular energy, the energy of the molecule with respect to a zero in which all of the nuclei and electrons are separated to infinity. It is possible to calculate the first derivatives (gradients) of this total molecular energy with respect to displacements of the nuclei, and geometries at which the gradients vanish (and the second derivatives are all positive) are termed true minimum energy structures. This process is known as geometry optimisation, and the results of such calculations are routinely compared with experimentally determined geometric structures (as obtained, for example, from X-ray or electron diffraction techniques).
The absolute values of HF-SCF total energies are vast, typically many hundreds of atomic energy units (1 a.u. (Hartree) ≡ 2625 kJ mol−1). Crucially, however, total HF-SCF energies do not contain a contribution from the effects of electron correlation (the tendency of electrons to avoid each other owing to their charged nature) as the HF-SCF method is intrinsically incapable of describing these effects. Correlation energies are tiny fractions of the total (of the order of a few hundred kJ mol−1) yet are very significant at a chemical level (comparable to chemical bond energies) and usually the most time-consuming part of an ab initio calculation lies in the extension of the HF-SCF method to include electron correlation. These so-called ‘post Hartree–Fock’ methods are broadly divided into two; perturbation approaches such as Møller–Plesset theory (the MPn techniques) and methods based on configuration interaction (including coupled cluster methods such as CCSD(T)).
The ab initio methods discussed thus far are entirely non-relativistic, and might therefore be considered unsuitable for the study of actinide systems. Relativistic analogues of HF theory do exist, but these Dirac–Fock techniques are difficult to implement computationally and are very demanding in terms of computer resources, and are not yet routinely used on 5f element systems. Rather, the effects of relativity are usually incorporated into ab initio calculations through the basis set. Relativistic basis sets are derived from (usually) Dirac–Fock calculations on individual atoms, and these relativistically-modified atomic functions are then used in ab initio molecular calculations. It should also be noted that most ab initio calculations on actinide compounds make use of relativistic pseudopotential (or relativistic effective core potential (RECP)) basis sets. These replace the computationally demanding yet chemically unimportant core electrons by functions designed to mimic their effects on the valence electrons, which are themselves treated explicitly.
The fundamental quantity of DFT is the charge density, from which (according to Hohenberg and Kohn) all of the ground state properties of a system may be derived. Kohn and Sham used mathematical sleight of hand to recast the solution to density functional problems in terms of quantities that are straightforward to evaluate, with the exception of one—the exchange-correlation energy of a system with a given charge density. The principal drawback to density functional methods was and is that the correct mathematical form of the exchange-correlation energy is not known, although it has been the subject of intense research for many years and very good approximate formulations are now available.
There are both similarities and differences between DFT and ab initio methods and their computational implementations. We have already seen that a serious flaw in the HF-SCF approach is its inability to include electron correlation. By contrast, even simple formulations of DFT can account for correlation effects in some way. However, while the theoretical pathway to including all correlation effects in ab initio calculations is well known,† there is no such clear way forward for DFT.
The computational time required for HF-SCF calculations formally scales as N4, where N is the number of basis functions. Post HF calculations are even more demanding, up to N7 for certain methodologies. In practice, this places severe restrictions on the size of the problem that can be tackled with ab initio methods, although ever increasing computer power is continually moving the goalposts. By contrast, most computational implementations of DFT scale somewhere between N2 and N3 in the number of basis functions (molecular DFT codes typically use atom-centred basis functions similar to those employed in ab initio methods), making DFT calculations much cheaper computationally than HF and particularly post HF approaches. Furthermore, ab initio methods are in practice restricted to Gaussian basis sets (for reasons of computational feasibility), whereas it is much more straightforward to use Slater basis functions in DFT calculations.‡ The significance of this is that in general the mathematical description of AOs requires fewer Slater functions than Gaussian functions, meaning that N in DFT calculations can be smaller than in analogous ab initio methods, further increasing the relative speed of DFT.
The speed advantages of DFT are clearly of enormous importance in actinide chemistry. Furthermore, the speed of DFT methods is such that the inclusion of relativistic effects is not restricted to the basis sets; relativistic Hamiltonians have been formulated for density functional methods and are now routinely employed in actinide calculations.
3 Case studies
3.1 π Backbonding in uranium complexes
The Dewar–Chatt–Duncanson model of synergic bonding is now more than 50 years old,12 and has gained widespread acceptance amongst inorganic and organometallic chemists during this period. The principal feature of the model is that a ligand, e.g. CO or η2-C2H4, binds to a transition metal in two complementary ways; donation of σ electron density from a filled ligand orbital into an empty metal function, with simultaneous backdonation of metal electron density into vacant (and usually antibonding) ligand levels. This backdonation is of π symmetry with respect to the metal–ligand bonding axis. This model elegantly rationalises many experimental observations, e.g. the lengthening of the CO bond and reduction in the CO stretching frequency upon coordination of CO to a transition metal, as a result of partial population of the CO π* orbital.In 1986, Brennan et al. reported the synthesis and characterisation of [U Cp‴3CO] (Cp‴ = η5-C5H4SiMe3),13 and noted that the CO stretching frequency was some 170 cm−1 lower than in free CO. Subsequent DFT calculations on the model complex [UCp3CO]14 indicated that the CO was functioning as a classic Dewar–Chatt–Duncanson ligand to the actinide centre. Thus, computational evidence was found for σ donation from the 5σ highest occupied molecular orbital (HOMO) of CO into the vacant uranium 6dz2 orbital, in conjunction with significant π backbonding from the uranium 5fπ AOs into the vacant CO π* levels. This result neatly explained the experimental infra-red data, and provided further evidence that the early actinides share certain similarities in their chemistry with the transition metals.1
Dinitrogen (N2) is isoelectronic with CO, and might therefore be expected to interact in a broadly similar manner with metal centres. Indeed, virtually all transition metals have been found to form complexes with N2, and many hundreds of such systems have been characterised. Furthermore, the coordination mode of the N2 in the vast majority of these complexes is end-on and linear along the M–N–N vector, analogous to the C-bound orientation of the CO ligand in its transition metal complexes.
By contrast to the wealth of dinitrogen transition metal chemistry, only three dinitrogen actinide complexes have been reported.15–17 All three are bimetallic uranium systems (that of Cummins et al. being a mixed U/Mo compound), and the first to be reported was [{(NN′3)U}2(μ2-η2:η2-N2)] [NN′3 = N(CH2CH2NSiButMe2)3] shown schematically in Fig. 1a. This beautiful molecule features side-on coordination of the N2 to both uranium centres. Interestingly, the N–N distance is essentially the same as in free dinitrogen, prompting Roussel and Scott to suggest that the N2 binds to the uranium atoms in a σ fashion—the donor orbital being the filled N2 πu N–N bonding level—and that each [UNN′3] unit is functioning as a very potent Lewis acid. This bonding mode is represented schematically in Fig. 1b.
![Schematic representation of (a) the molecular structure of [{(NN′3)U}2(μ2-η2:η2-N2)] [(NN′3 = N(CH2CH2NSiButMe2)3] and (b) the bonding in the U2N2 core as suggested by Roussel and Scott.15](/image/article/2003/CS/b204253n/b204253n-f1.gif) | ||
| Fig. 1 Schematic representation of (a) the molecular structure of [{(NN′3)U}2(μ2-η2:η2-N2)] [(NN′3 = N(CH2CH2NSiButMe2)3] and (b) the bonding in the U2N2 core as suggested by Roussel and Scott.15 | ||
Roussel and Scott’s suggestion, while entirely reasonable, was almost immediately called into question by our DFT study of the electronic structure of the model complex [{(NH2)3(NH3)U}2(μ2-η2:η2-N2)].18 These calculations indicated that the only significant metal/N2 interaction is π backbonding from the 5f AOs of the formally U(III) centres into the N2 πg N–N antibonding MOs. A three-dimemsional representation of one of the two such π backbonding MOs is shown in Fig. 2. It would therefore appear that [{(NH2)3(NH3)U}2(μ2-η2:η2-N2)] (and presumably [{(NN′3)U}2(μ2-η2:η2-N2)]) contains only the π backbonding part of the more typical synergic bond, the lack of σ donation most likely arising because the N2 πu levels lie too low in energy to function as effective donors.
![Three-dimensional representation of the one of the two U2
→ N2
π backbonding MOs in [{(NH2)3(NH3)U}2(μ2-μ2:η2-N2)], a computational model for [{(NN′3)U}2(μ2-η2:η2-N2)]. Figure from ref. 18.](/image/article/2003/CS/b204253n/b204253n-f2.gif) | ||
| Fig. 2 Three-dimensional representation of the one of the two U2 → N2 π backbonding MOs in [{(NH2)3(NH3)U}2(μ2-μ2:η2-N2)], a computational model for [{(NN′3)U}2(μ2-η2:η2-N2)]. Figure from ref. 18. | ||
As noted above, one of the most curious features of [{(NN′3)U}2(μ2-η2:η2-N2)] is the N–N bond length. The significant population of the N2 πg level predicted by the DFT calculations on the model system should result in an increase in the N–N distance (in the same way that the CO bond is almost always longer in transition metal carbonyl complexes than in free CO), and indeed geometry optimisations of [{(NH2)3(NH3)U}2(μ2-η2:η2-N2)] always produce a shortening of the U–N(N2) distance and a lengthening of the N–N distance in comparison with experiment. In an attempt to get to the bottom of this issue, we conducted further calculations on UN2 and U2N2.19,20 However, the conclusions from these studies were essentially the same as the original study, i.e. that uranium → N2 π backdonation is the dominant interaction in the side-bound systems and that the optimised N–N distances are significantly longer than in free N2. We therefore suggested that the most probable explanation20 for the discrepancy between theory and experiment is that the NN′3 ligands of the experimental system are so bulky that they prevent the two ends of the molecule from coming any closer together, as would accompany a reduction in the U–N(N2) distance. Fig. 3 presents a space-filling diagram of [{(NN′3)U}2(μ2-η2∶η2-N2)], which shows the way in which the SiMe2tBu groups on the two ends of the molecule interlock, preventing closer approach of the uranium atoms at the core of the complex. Thus in both the real system and its computational models there is an electronic driving force toward U–N(N2) shortening and N–N lengthening, but in the real system this is opposed (and overcome) by the highly sterically demanding NN′3 ligands.
![Space-filling diagram of [{(NN′3)U}2(μ2-η2:η2-N2)] [(NN′3 = N(CH2CH2NSiButMe2)3]. Reprinted with permission from ref. 20. Copyright (2001) Elsevier Science.](/image/article/2003/CS/b204253n/b204253n-f3.gif) | ||
| Fig. 3 Space-filling diagram of [{(NN′3)U}2(μ2-η2:η2-N2)] [(NN′3 = N(CH2CH2NSiButMe2)3]. Reprinted with permission from ref. 20. Copyright (2001) Elsevier Science. | ||
Very recent experimental work by Cloke and Hitchcock has, however, called this suggestion into question.17 These workers report a dinitrogen uranium complex featuring both Cp* and pentalene ancillary ligands, [(U(η5-C5Me5)(η8-C8H4{SiiPr3-1,4}2)2(μ-η2:η2-N2)] shown both schematically and as an ORTEP in Fig. 4. By contrast to [{(NN′3)U}2(μ2-η2:η2-N2)] the N–N distance in the Cloke system is significantly longer than in free N2, consistent with the presence of an N–N double bond (i.e. the dinitrogen ligand is best formulated as N22−). Intriguingly, however, the U–N distances are essentially the same as in [{(NN′3)U}2(μ2-η:η2-N2)], and Cloke has suggested that the difference between [{(NN′3)U}2(μ2-η2:η2-N2)] and [(U(η5-C5Me5)(η8-C8H4{SiiPr3-1,4}2)2(μ-η2:η2-N2)] may be a consequence of different frontier orbital geometries in the two ligand environments. Further theoretical work is required (and indeed underway) on these intriguing compounds.
![Schematic (upper) and ORTEP (lower—iPr3 groups omitted for clarity) diagrams of [(U(η5-C5Me5)(η8-C8H4{SiiPr3-1,4}2)2(μ-η2:η2-N2)]. Reprinted with permission from ref. 17. Copyright (2002) American Chemical Society.](/image/article/2003/CS/b204253n/b204253n-f4.gif) | ||
| Fig. 4 Schematic (upper) and ORTEP (lower—iPr3 groups omitted for clarity) diagrams of [(U(η5-C5Me5)(η8-C8H4{SiiPr3-1,4}2)2(μ-η2:η2-N2)]. Reprinted with permission from ref. 17. Copyright (2002) American Chemical Society. | ||
One of the principal results from the computational studies of both [UCp3CO] and [{(NH2)3(NH3)U}2(μ2-η2:η2-N2)] is that formally U(III) centres are more than capable of functioning as π bases, and very recent work by Mazzanti et al. has further reinforced this conclusion.21 These workers have conducted DFT calculations on [M(pyrazine)I3] (Fig. 5a), [M(acetonitrile)I3] (Fig. 5b), and [M(pyrazine)3I3] (M = La, Nd, U), models for experimentally characterised tris[(2-pyrazinyl)methyl]amine systems. Geometry optimisations of these compounds reproduce experimental trends, i.e. there is a reduction in r(M–N) from lanthanum to uranium even though the ionic radii of La3+ and U3+ are very similar. The calculations reveal that there is essentially no orbital interaction between Ln3+ and the N-donor ligands, by contrast to the actinide system in which there is π backdonation from the 5f AOs of U3+ into the π* levels of both acetonitrile and pyrazine.
![Schematic diagrams of (a) [U(pyrazine)I3] and (b) [U(acetonitrile)I3], studied by Mazzanti et al.21](/image/article/2003/CS/b204253n/b204253n-f5.gif) | ||
| Fig. 5 Schematic diagrams of (a) [U(pyrazine)I3] and (b) [U(acetonitrile)I3], studied by Mazzanti et al.21 | ||
3.2 Bis benzene complexes
So-called sandwich molecules, in which two planar and parallel carbocyclic rings sandwich a metal centre, occupy a fundamental position in organometallic chemistry. Indeed, the archetypal actinide compound is arguably the bis cyclooctatetraene (bis ‘COT’) system—the ‘actinocenes’—in which a formally An(IV) centre (An = generic actinide) is sandwiched between two formally dianionic η8-C8H8 rings. These systems, which are unique to the f-block, have been the subject of numerous experimental and theoretical studies since the early 1960s. However, since there have been no computational contributions in this area since the mid 1990s, I do not intend to discuss the actinocenes here. Rather, I refer the reader to the excellent summary of Dolg and Fulde,22 and move on to describe more recent computational work on the less well studied, but equally fascinating, bis benzene complexes of the actinides.Bis benzene complexes are, of course, well known among the transition elements (with those of the group 6 metals being perhaps the best examples), but are much less common in the f-block. Indeed, although bis η6-arene (in particular η6-1,3,5-C6H3tBu3 ‘TTB’) complexes of the lanthanides have been known for more than a decade, no neutral actinide bis benzene system has yet been synthesised. In 1999 Hong et al. published the results of an in-depth ab initio study of the energetics and bonding in [MBz2] (M = La, Ce, Gd, Tb, Lu, Th and U; Bz = η6-C6H6).23 These calculations were performed with several aims, including determining if bis benzene actinide compounds are intrinsically unstable or if they should in principle be isolable, and also to gain insight into the main metal–ring bonding mechanisms. They employed extensive electron correlation techniques (e.g. state-averaged CASSCF, MRCI and CCSD(T) methods) in conjunction with large pseudopotential basis sets, and found that both [ThBz2] and [UBz2] are stable with respect to dissociation into two benzene rings plus the metal (metal–ring bonding energies of 167 and 175 kJ mol−1 respectively for the thorium and uranium compounds were calculated at the CCSD(T) level of theory), and hence should in principle be isolable experimentally. Hong et al. noted that the An–Bz interaction is weaker than in analogous bis COT systems due to the minimal ionic contributions to the bonding (benzene is of course formally neutral and closed shell, and no formal electron transfer from metal to ligands takes place on complex formation, unlike in COT or indeed cyclopentadienyl systems).
Hong et al. also probed the nature of the metal–ring interaction. They found that the dominant bonding interaction is in some ways reminiscent of [{(NH2)3(NH3)U}2(μ2-η2:η2-N2)] discussed above, in that it involves backdonation from metal to ligand. The details of the interaction are rather different, however, in that in [AnBz2] the filled actinide orbitals are primarily the 6d±2,§ not 5f as in the case of [{(NH2)3(NH3)U}2(μ2-η2:η2-N2)], and the interaction is of δ symmetry with respect to the metal–ring centroid axes. The acceptor levels on the ligands are the empty π2 orbitals. These are MOs of π symmetry in free benzene, and are formed from the C 2p AOs which lie perpendicular to the molecular plane. The ‘2’ subscript indicates that these π MOs have two vertical nodes (i.e. nodes perpendicular to the plane of the benzene ring).24 It is worth noting that the conclusions concerning the main metal–ring bonding in [AnBz2] are not dissimilar to those from earlier experimental studies on [CrBz2] and [Mo(η6-C6H5Me)2],25 although the calculations of Hong et al. also indicate a small 5f±2 orbital involvement in the δ backbonding in [ThBz2] and [UBz2], which is of course not present in the transition metal systems.
This research took an interesting twist shortly after Hong et al.’s contribution was published. Hong et al. had assumed in their calculations that the most stable geometry of [LnBz2] and [AnBz2] is with the rings parallel to one another, i.e. the molecules belong to the D6h point group. This is hardly unreasonable, given that this is the experimentally determined geometry of the transition metal analogues, and also of [Gd(TTB)2].26 However, this assumption was almost immediately called into question by Li and Bursten, who used DFT methods to probe the geometric structures of [AnBz2] (An = Th–Am) and [An(η6-C6H3R3)2] (An = Th, U, Pu; R = Me, tBu).27 They found that the most stable geometry of [AnBz2] is significantly bent, i.e. the angle subtended by vectors connecting the ring centroids and the actinide centre is substantially less than 180° (ranging from 135–142°, depending on the metal). This bending is electronically driven, with greater covalency in the bent structure arising from greater Bz → An 6d and 5f donation.
Replacement of three H atoms on each ring by Me groups also results in bent structures. However, when the Me groups are replaced by the much bulkier tBu units the most stable geometry reverts to linear, i.e. for very bulky R substituents the steric repulsion between the rings overcomes the electronic preference for bending. These conclusions are summarised in Fig. 6.
![Schematic summary of the geometric structures of [An(η6-C6H3R3)2] (An = Th, U, Pu; R = Me, tBu), as suggested by Li and Bursten.27](/image/article/2003/CS/b204253n/b204253n-f6.gif) | ||
| Fig. 6 Schematic summary of the geometric structures of [An(η6-C6H3R3)2] (An = Th, U, Pu; R = Me, tBu), as suggested by Li and Bursten.27 | ||
Shortly after Li and Bursten’s work was published, Hong et al. revisited [ThBz2] and [UBz2] using both ab initio and DFT methods.28 They found that neither molecule is bent at the ab initio HF–SCF level. Inclusion of electron correlation, however, using MP2 and CCSD(T) methods, results in significantly bent structures (c. 140–145°), although the energy differences between the linear and bent geometries are so small that the most stable geometry cannot be unequivocally established using these techniques. Hong et al. then conducted DFT studies which also indicated that the bent structures are the most stable, and finally concluded that ‘qualitatively....we confirm their [Li and Bursten’s] result that the bis benzene complexes of thorium and uranium have bent structures’, and that bending enhances metal–ring bonding.
3.3 The valence electronic structure of the uranyl ion
The uranyl ion, UO22+, is ubiquitous in uranium chemistry on account of its high chemical stability. Indeed, Zhang and Pitzer29 pointed out that about half of all the known uranium compounds contain the uranyl ion, and this central position has prompted extensive theoretical and experimental research into its electronic structure. This interest is also motivated by the contrast between UO22+ and transition metal analogues such as MoO22+, for while the latter are often bent, UO22+ is invariably linear,¶ with a much shorter (and stronger) U–O bond than would be expected from the M–O distances in the transition metals.An excellent review of uranyl (and other actinyl) electronic structural work to 1992 was provided by Denning,30 and Zhang and Pitzer gave a much briefer but equally well written summary in 1999.29 In this section I shall present our current understanding of the valence electronic structure of UO22+, including information drawn from theoretical and experimental studies performed in the last three years. A clear and consistent picture is emerging of the bonding in this fundamental system.
UO22+ has a closed shell singlet ground state with 12 valence electrons (originating in the oxygen 2p and uranium 5f, 6d and 7s AOs). These valence electrons are accommodated in four MOs, transforming as πg, πu, σg and σu in the D∞h molecular point group, and the centrosymmetric nature of this point group neatly separates the contributions to these MOs of the uranium 6d (g) and 5f (u) AOs. The relative ordering and compositions of these four MOs has been the subject of debate for decades. In 2000, I reported the results of relativistic DFT calculations on UO22+,31 and the relative energies of the four valence MOs taken from this work are shown in Fig. 7. This figure shows the results of two calculations; one (left) in which the uranium 6p AOs are allowed to participate in the valence electronic structure and the second (right) in which these AOs are placed into the uranium frozen core (i.e. orbitals which are not altered from their atomic values in the molecular calculation). I shall return to this distinction later, after discussing another feature of Fig. 7.
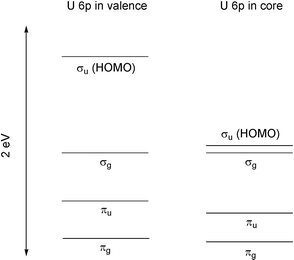 | ||
| Fig. 7 Relative energy levels of the valence MOs of UO22+, taken from DFT calculations31 in which the uranium 6p AOs are allowed to participate in the valence electronic structure (left) and placed in the uranium frozen core (right). The energy of the σg MO is arbitrarily set to be equal in the two diagrams. | ||
My results show (by contrast to several ab initio studies, as summarised in reference 31) that both of the π MOs are more stable than the σ levels, suggesting that π bonding is stronger than σ. This is contrary to familiar assumptions about the relative extents of σ and π overlap, but is the MO ordering suggested by Denning in 1992 on the basis of the then-available experimental data.30 It is pleasing to report that very recent oxygen-Kα X-ray absorption spectroscopy (XAS) work by Denning et al.32 clearly supports the πg < σg ordering, on the grounds that the unoccupied πg* antibonding counterpart of the πg valence MO lies about 2 eV above the σg* level. Taken together, the theoretical and experimental data clearly indicate that, for the g type MOs at least, U–O π bonding is more significant than σ.
Returning to Fig. 7, it is clear that the inclusion of the uranium 6p semi-core AOs in the molecular SCF procedure makes very little difference to the relative energy of the πg, πu, and σg MOs (or, indeed, to their absolute eigenvalues). By contrast, it has a pronounced destabilizing effect on the σu level, and the involvement of a semi-core level in this manner has been termed the ‘pushing from below’ mechanism.33 The σu HOMO has a contribution from the uranium 5fσ AO, which is bonding with the oxygen 2pσ AOs. However, the uranium 6pσ/oxygen 2pσ interaction in the σu HOMO is U–O antibonding, and hence the net result of the semi-core 6pσ admixture into the σu HOMO is to reduce its U–O bonding properties significantly, and destabilise it relative to the other valence MOs.
Denning et al.’s recent experimental study32 used X-ray emission spectroscopy (XES) in addition to the XAS measurements described above. The XES experiments detected a significant oxygen 2p contribution to the highest energy component (σ) of the uranium 6p semi-core orbitals (the next filled MO below the πg of Fig. 7, though well separated in energy). This result provides powerful evidence for the involvement of the uranium 6pσ AO in the σu HOMO, which may be viewed as the uranium 6p/oxygen 2p antibonding partner of the mainly uranium 6pσ based level shown by XES to possess oxygen 2p character.
The involvement of the 6pσ semi-core level in the σu HOMO results in a partial hole in the 6p shell. Estimates vary, but computational studies29,31,34,35 typically yield a 6p population of c. 5.5 (reduced from the purely atomic value of 6.0) and find that this hole is almost entirely in the 6pσ level.
Denning et al.’s XES data also suggest that, while the πg, πu, and σg MOs are predominantly of oxygen 2p character, the σu orbital contains only a small contribution from the oxygen atoms. My DFT analysis indicated a 34% oxygen 2p contribution to the σu HOMO,31 rather larger than experiment would suggest. I note, however, that the calculated oxygen 2p content of the πg, πu, and σg MOs is 64% or greater, and hence that qualitatively at least the DFT calculations concur with experimental work in finding a much reduced oxygen 2p contribution to the σu HOMO.
In summary, it is now clear that the valence MO structure of UO22+ features three closely-spaced levels of predominantly oxygen 2p character (πg, πu, σg), of which the πg is the most U–O bonding. The fourth MO—the σu HOMO—differs from the other three in that it (a) has much less oxygen 2p character (b) is significantly less stable and (c) is less bonding between the uranium and the oxygen. All of these features of the σu level can be attributed to its admixture of uranium semi-core 6pσ.
Before leaving the uranyl ion, I would like to mention briefly the work of Schreckenbach et al. which raises the intriguing possibility of the existence of ‘cis-uranyl’ compounds.3,36 As I indicated at the start of this section, the known compounds of UO22+ all feature an essentially linear uranyl unit. However, Schreckenbach et al.’s DFT work on [UO2X4]2− (X = OH, F, Cl) has revealed that local minimum structures with a significantly bent O–U–O unit (angles in the region of 110–135°) lie only c. 75 kJ mol−1 higher in energy than the global, linear minimum. Furthermore, these workers note that all of the bent uranyl conformers of [UO2(OH)4]2− have permanent dipole moments,36 by contrast to some of the linear structures, and hence suggest that polar solvents should stabilise the “cis-uranyl” conformers relative to the linear. To my knowledge, however, no compound featuring a bent uranyl unit has yet been reported.
3.4 The trans influence and inverse trans influence
The trans influence is well established in molecular transition metal chemistry, and is standard material in undergraduate courses and textbooks. It is typically associated with square planar and pseudo-octahedral systems, in which certain ligands (e.g. H−, O2−, N3−) selectively weaken and lengthen the bonds trans to their own position. There have many attempts to explain the trans influence, dating back to 1935, when Grinberg suggested that the trans-directing ligand polarises the metal such that negative charge builds up in the trans position. Repulsion between this negative charge and that of the trans ligand results in a lengthening of the metal–trans ligand bond. Subsequent refinements of this theory have invoked the use of ns and/or (n − 1)d orbitals on the metal, in what amounts to a recasting of Grinberg’s argument in MO terms.37In 1995 Lyne and Mingos used DFT methods to probe the origin of the trans influence in pseudo-octahedral [OsNCl5]2−.38 These workers focussed on the valence MO structure, and found that the second HOMO of this system is π bonding between the metal and the nitrido group but π antibonding between the Os and the cis chlorines. Increasing the N–Os–Clcis angle above 90° causes this orbital to become more Os–N bonding and less Os–Clcis antibonding, i.e. there is an orbital driving force toward increased N–Os–Clcis angle. Lyne and Mingos also found two other orbitals which oppose this bending (i.e. which are destabilised as the N–Os–Clcis angle increases). The net result of these electronic factors is an N–Os–Clcis angle of 96°.
The consequence of this increased N–Os–Clcis angle is increased non-bonded repulsions between the cis and trans chlorine atoms, which result in a 0.2 Å lengthening of the Os–Cltrans bond, i.e. a trans influence. This process is summarised in Fig. 8. Our subsequent studies on mer-[Ti(NR)Cl2(NH3)3] (R = But, C6H5 and 4-C6H4NO2) found that this mechanism also contributes to the trans influence in these more complex systems, although it is not the sole source of the Ti–(NH3)trans lengthening.39
![Schematic diagram of the origin of the inverse trans influence in [OsNCl5]2−.38 Note that, for clarity, only two of the four cis chlorine atoms are shown.](/image/article/2003/CS/b204253n/b204253n-f8.gif) | ||
| Fig. 8 Schematic diagram of the origin of the inverse trans influence in [OsNCl5]2−.38 Note that, for clarity, only two of the four cis chlorine atoms are shown. | ||
By contrast to the regular trans influence typical of transition metal systems, certain pseudo-octahedral actinide complexes display the opposite trend in cis and trans bond lengths. A good example is [UOCl5]−, in which the U–Cltrans distance is 0.1 Å shorter than the U–Clcis.40 In 1992 Denning coined the phrase inversetransinfluence (ITI) to describe this effect,30 and suggested a ‘naive, but pleasingly simple’ explanation for its origin. In an argument reminiscent of UO22+, Denning proposed the indirect involvement of the uranium 6p semi-core AOs in the metal–ligand interactions. Specifically, polarisation of the metal’s core by the tightly bound ligand (in the case of [UOCl5]− the oxo group) leads to a quadrupolar charge distribution due to interaction of the highest occupied core orbitals (the 6p levels) with the formally empty valence 5f orbitals. This quadrupolar charge distribution builds up negative charge in the cis position relative to the trans, and the electrostatic interaction of this non-spherical charge distribution with the anionic chlorides produces the ITI.
Crucially, the relative parity of the (semi-)core and valence functions is the same in the case of [UOCl5]−. Contrast this with the (early) transition metals, for which the core (p) and valence (d) orbitals have the opposite parity. Under these circumstances the core polarisation is predominantly dipolar, leading to a regular trans influence via a mechanism not dissimilar to that first proposed by Grinberg.
We recently explored the ITI in [AnOX5]n−(An = Pa, n = 2; An = U, n = 1; An = Np, n = 0; X = F, Cl or Br) using DFT methods.41 Comparative calculations in which the metal’s 6p orbitals were either frozen at their atomic values or freed up to participate in the valence electronic structure indicated that the ITI can be only partly explained by Denning’s suggestion, i.e. an (albeit reduced) ITI was found in all of the complexes even with the metal’s 6p AOs placed in the frozen core. We therefore concluded that there must be at least one other factor contributing to the ITI, and turned to an orbital analysis similar to that presented by Lyne and Mingos on [OsNCl5]2−.
Taking [UOBr5]− as a representative system, we analysed the changes in the energies of the valence MOs as a function of O–U–Brcis angle. This revealed only three MOs whose energies change by > ±7 kJ mol−1 as the angle is increased from 90° to 102°. Two of these orbitals are significantly destabilised (22 kJ mol−1 and 55 kJ mol−1) and analysis showed them to be the equivalent orbitals of those identified by Lyne and Mingos as opposing the increase in N–Os–Clcis angle in [OsNCl5]2−. The third orbital of [UOBr5]− is stabilised by the bending process, but only by 13 kJ mol−1, and hence we concluded that there is not an orbital driving force for the O–An–Xcis angle to exceed 90°, by contrast to the transition metal system. Indeed, the analysis of [UOBr5]− suggested that there is a reasonably strong preference for the O–U–Brcis angle to be 90°, in agreement with the fully optimised geometries of all nine [AnOX5]n− in which the O–An–Xcis angles deviate from 90° by no more than 1.7° (with the exception of [PaOF5]2−, for which it is 93°).
The lack of an orbital driving force for greater-than-90° O–An–Xcis angles goes a long way to explaining the lack of a trans influence in the actinide systems, as there will not be the non-bonded Xcis/Xtrans repulsions identified in [OsNCl5]2− (or at most they will be much reduced). Three questions still remain, however. Are there any orbital reasons why An–Xcis should be longer than An–Xtrans, is there an actinide equivalent of the key [OsNCl5]2− orbital (that shown in Fig. 8) and, if so, why does it not provide the same driving force toward bending at the metal?
The answer to all three questions is almost certainly tied up with the 9a1 MO of all of the actinide systems studied. A three-dimensional representation of this orbital in [NpOF5] is shown in Fig. 9, from which it may be seen that the orbital is σ bonding along the O–Np–Ftrans vector, but π antibonding between the metal and the cis halogens. While the relative extents of these interactions vary from system to system, the nature of the orbital does not. The An–Xcis π antibonding (and, to a lesser extent, the An–Xtrans σ bonding) nature of this orbital is most likely the origin of the residual ITI (i.e. after the 6p effects have been eliminated) in the actinide systems.
![Three-dimensional representation of the 9a1 MO of [NpOF5], from ref. 41. The molecule is oriented with the oxygen atom to the left.](/image/article/2003/CS/b204253n/b204253n-f9.gif) | ||
| Fig. 9 Three-dimensional representation of the 9a1 MO of [NpOF5], from ref. 41. The molecule is oriented with the oxygen atom to the left. | ||
In [OsNCl5]2−, the Os–Clcis π antibonding MO is significantly stabilised on increasing the N–Os–Clcis angle. This is not the case for the 9a1 level of the actinide systems (e.g. the 9a1 MO of [UOBr5]− is stabilised by only 5 kJ mol−1 as the O–U–Brcis angle increases from 90° to 102°). We suggested that the reason for the difference between the transition metal and the actinide systems lies in the nature of the metal AO involved in the M–Xcis π antibonding MO. Os–N dπ–pπ bonding is enhanced by increasing the N–Os–Clcis angle (Fig. 8). By contrast, the metal 5fz3 contribution to the 9a1 MO of the actinide systems means that this level is σ bonding between the metal and the oxo group (Fig. 9). Increasing the O–An–Xcis angle has little or no effect on the An–O σ bond, resulting in a much smaller stabilisation of this orbital and hence no overall orbital driving force toward bending.
4 Concluding remarks
I hope that in this review I have given the reader a flavour for the type of problem that is now being tackled in computational actinide chemistry. It is clear that computational chemists are bringing the same techniques and analysis tools to bear on the 5f elements that have been used for a long time in the s, p and d blocks. Improvements in computer power and in the coding of relativistic effects means that reliable computations on actinide systems are increasingly feasible, though not straightforward. This increased computational feasibility is important as the experimental study of most of the actinide elements is fraught with technical difficulties. I therefore suggest that computational techniques have a predictive role to play, and in the best cases could/should be used to minimise the number of experiments that must be done using the more radioactive and toxic 5f elements (e.g. neptunium and plutonium).I have heard it said that the actinides, particularly the early elements, are ‘big transition metals’. I would argue that this does not do justice to the often unique chemistry of these elements. Indeed, every example I have used in this review describes one or more actinide elements acting in a way which is different from the chemistry of analogous d-block systems. I look forward to continued challenges and surprises at the foot of the periodic table.
5 Acknowledgement
I am grateful to Andrea Sella for helpful discussions.6 References
-
N. Kaltsoyannis and P. Scott, The f elements, Oxford University Press, Oxford, 1999 Search PubMed
.
- M. Pepper and B. E. Bursten, Chem. Rev., 1991, 91, 719 CrossRef CAS
- G. Schreckenbach, P. J. Hay and R. L. Martin, J. Comp. Chem., 1999, 20, 70 Search PubMed
- P. Pyykkö, Chem. Rev., 1988, 88, 563 CrossRef CAS
- N. Kaltsoyannis, J. Chem. Soc., Dalton Trans., 1997, 1 RSC
-
F. Jensen, Introduction to Computational Chemistry, Wiley, Chichester, 1999 Search PubMed
- E. J. Baerends, W. H. E. Schwarz, P. Schwerdtfeger and J. G. Snijders, J. Phys. B, 1990, 23, 3225 Search PubMed
- N. Kaltsoyannis and B. E. Bursten, Inorg. Chem., 1995, 34, 2735 CrossRef CAS
-
P. W. Atkins and R. S. Friedman, Molecular Quantum Mechanics, Oxford University Press, Oxford, Third edn., 1997 Search PubMed
-
A. Szabo and N. S. Ostlund, Modern Quantum Chemistry, McGraw-Hill, New York, 1989 Search PubMed
-
R. G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules, OUP, Oxford, 1989 Search PubMed
- D. M. P. Mingos, J. Organomet. Chem., 2001, 635, 1 CrossRef CAS
- J. G. Brennan, R. A. Andersen and J. L. Robbins, J. Am. Chem. Soc., 1986, 108, 335 CrossRef CAS
- B. E. Bursten and R. J. Strittmatter, J. Am. Chem. Soc., 1987, 109, 6606 CrossRef CAS
- P. Roussel and P. Scott, J. Am. Chem. Soc., 1998, 120, 1070 CrossRef CAS
- A. L. Odom, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 1998, 120, 5836 CrossRef CAS
- F. G. N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 2002, 124, 9352 CrossRef CAS
- N. Kaltsoyannis and P. Scott, Chem. Commun., 1998, 1665 RSC
- K. L. Brown and N. Kaltsoyannis, J. Chem. Soc., Dalton Trans., 1999, 4425 RSC
- P. Roussel, W. B. Errington, N. Kaltsoyannis and P. Scott, J. Organomet. Chem., 2001, 635, 69 CrossRef CAS
- M. Mazzanti, R. Wietzke, J. Pécaut, J.-M. Latour, P. Maldivi and M. Remy, Inorg. Chem., 2002, 41, 2389 CrossRef CAS
- M. Dolg and P. Fulde, Chem. Eur. J., 1998, 4, 200 CrossRef CAS
- G. Hong, F. Schautz and M. Dolg, J. Am. Chem. Soc., 1999, 121, 1502 CrossRef CAS
-
F. A. Cotton, Chemical Applications of Group Theory, Wiley-Interscience, New York, Third edn., 1991 Search PubMed
- J. G. Brennan, G. Cooper, J. C. Green, N. Kaltsoyannis, M. A. MacDonald, M. P. Payne, C. M. Redfern and K. H. Sze, Chem. Phys., 1992, 164, 271 CrossRef CAS
- J. G. Brennan, F. G. N. Cloke, A. A. Sameh and A. Zalkin, J. Chem. Soc., Chem. Commun., 1987, 1668 RSC
- J. Li and B. E. Bursten, J. Am. Chem. Soc., 1999, 121, 10243 CrossRef CAS
- G. Hong, M. Dolg and L. Li, Int. J. Quant. Chem., 2000, 80, 201 CrossRef CAS
- Z. Zhang and R. M. Pitzer, J. Phys. Chem. A, 1999, 103, 6880 CrossRef CAS
- R. G. Denning, Struct. Bonding, 1992, 79, 215 Search PubMed
- N. Kaltsoyannis, Inorg. Chem., 2000, 39, 6009 CrossRef CAS
- R. G. Denning, J. C. Green, T. E. Hutchings, C. Dallera, A. Tagliaferri, K. Giarda, N. B. Brookes and L. Braicovich, J. Chem. Phys., 2002, 117, 8008 CrossRef CAS
- C. K. Jørgensen and R. Reisfeld, Struct. Bonding, 1982, 50, 121 Search PubMed
- W. A. de Jong, L. Visscher and W. C. Nieuwpoort, J. Mol. Struct. (THEOCHEM), 1999, 458, 41 CrossRef CAS
- S. Matsika and R. M. Pitzer, J. Phys. Chem. A, 2001, 105, 637 CrossRef CAS
- G. Schreckenbach, P. J. Hay and R. L. Martin, Inorg. Chem., 1998, 37, 4442 CrossRef CAS
- For a summary of these arguments, see
J. E. Huheey, E. A. Keiter and R. L. Keiter, Inorganic Chemistry, Principles of Structure and Reactivity, HarperCollins, New York, Fourth edn., 1993 Search PubMed
- P. D. Lyne and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 1995, 1635 RSC
- N. Kaltsoyannis and P. Mountford, J. Chem. Soc., Dalton Trans., 1999, 781 RSC
- J. F. de Wet and J. G. H. du Preez, J. Chem. Soc., Dalton Trans., 1978, 592 RSC
- E. O′Grady and N. Kaltsoyannis, J. Chem. Soc., Dalton Trans., 2002, 1233 RSC
Footnotes |
| † The method is known as full configuration interaction, and in the limit of infinite basis set size will recover all of the electron correlation energy. However, this method is computationally so demanding as to be restricted in practice to only the smallest molecules. |
| ‡ The primary difference between the two types of function is that the radial dependence of Gaussian functions contains a term in e−αr2, while Slater functions depend on e−αr. |
| § These are the 6dx2−y2 and 6dxy orbitals, in an axis system in which the z axis is the principal molecular (6-fold) rotation axis. |
| ¶ The wide variety of uranyl compounds is largely a function of the groups that coordinate to the uranium in the equatorial plane. |
| This journal is © The Royal Society of Chemistry 2003 |