The electronic structure of Cu+, Ag+, and Au+ zeolites
Gion
Calzaferri
*a,
Claudia
Leiggener
a,
Stephan
Glaus
a,
David
Schürch
a and
Ken’ichi
Kuge
b
aDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3000, Bern 9, Switzerland
bFaculty of Engineering, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba, 263, Japan
First published on 15th November 2002
Abstract
A variety of procedures have been used to prepare d10-zeolite materials. The electronic structure of these materials can be regarded to a first approximation as a superposition of the framework, of the charge compensating ions, of solvent molecules and of guest species. Zeolite oxygen to d10-ion charge transfer transitions dominate the electronic spectra if the ions coordinate to the zeolite oxygens. Specific coordination sites can influence the energy and the intensity of these transitions remarkably. Intra guest transitions dominate in quantum dot materials, as discussed in detail for luminescent Ag2S zeolite A. The zeolite is not needed for the photocatalytic water oxidation on Ag+/AgCl photo anodes with visible light. It can, however, be used to increase the active surface area substantially.
 Gion Calzaferri Gion Calzaferri | Gion Calzaferri (left) received his PhD degree at the University of Fribourg, Switzerland, 1971. He moved to the University of Berne, after a postdoc period at the University of Basel and physical-chemical research in connection with pharmacy at Ciba-Geigy Basel, Switzerland. He has been professor of physical chemistry in the Department of Chemistry and Biochemistry since 1988. His current research interests concern supramolecularly organized molecules, clusters and complexes in zeolite cavities, dye-zeolite materials as photonic antenna systems, and photochemical transformation and storage of solar energy. |
Claudia Leiggener (front, left) received her Diploma in Chemistry (2001) at the University of Berne. She is now working as a PhD student in Berne on time resolved spectroscopy of luminescent silver sulfide clusters. |
Stephan Glaus (middle) obtained his Diploma in Chemistry (1998) and his PhD degree (2002) from the University of Berne with a thesis entitled theoretical investigations of interfaces of silver and silver chloride clusters. |
David Schürch (right) studied chemistry at the University of Calgary and the University of Berne where he obtained his first degree. In 2002 he received his PhD degree at the University of Berne. |
Ken’ichi Kuge (front, right) has worked at the Department of Imaging Science, Chiba University, since 1979. He received his PhD degree from Kyoto University in 1984. He has been an associate professor for photosensitive materials since 1992. His current research interests concern photographic sensitivity, preparation of gold clusters and radiation measurement using photographic techniques. |
1 Introduction
The importance of host–guest materials in technology and environmental science can hardly be overemphasized. Basic research on such materials has been less attractive, probably because of the difficulties encountered in rigorous structure determination. Similar difficulties occur in some branches of nanoscience, which have attracted much fundamental research and also put new emphasis on host–guest materials. We are now learning how to describe the often fascinating properties of these materials in a more rigorous way and how to carry out experiments so that artifacts can be avoided.Zeolites are appealing host materials because they provide a large variety of well defined cavities and channels so that structural identification can concentrate on the shape and position of the guest, provided that care is taken that the framework of the host has not been affected by the synthesis procedure. This is usually easy to check by powder X-ray and vibrational spectroscopy. In this article we focus on the zeolites LTA, LTL, and FAU shown in Fig. 1. These zeolites are crystalline aluminosilicates with cavity and channel structures. Their lattice can be regarded as an enormous crystalline polyanion which contains cations for charge compensation.1
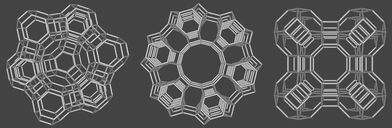 | ||
| Fig. 1 Framework of zeolite Y (left), zeolite L (middle), and zeolite A (right). The bridging oxygens are not shown. | ||
The electronic structure of a typical zeolite material can be regarded as a superposition of the electronic structure of the framework, of the charge compensating cations, of the solvent molecules and of the guest species. The band gap of about 7 eV and the HOMO position at about −10.7 eV of the zeolite framework are similar to those of α-quartz, despite the fact that the material is less dense.2–4
The charge compensating cations cause new electronic states, some of which may lie within the band gap region. They can interact with each other depending on their nature and the mean distance between them. The presence of solvent molecules, usually water, influences their interaction with the zeolite framework. The electronic structure of the zeolite framework and the charge compensating cations are not influenced by each other, as long as the solvation shell shields the cations from coordinating to zeolite oxygens. A partial removal of the solvent causes incomplete saturation of the coordination sites of the cations which, hence, try to compensate this by sticking closer to the zeolite oxygens. The involved states can be perturbed considerably which causes a change of the optical properties. Apart from charge compensating cations, guests can be present in neutral form, as molecules or as quantum dots or they can be occluded in anionic form. In these cases the electronic structure can be regarded in general as a superposition of the guest and the host with no or only little electronic interaction. However, vibrational interactions can be substantial. This is the underlying general understanding which will guide us in the description of d10-ion zeolites. We concentrate on Cu+, Ag+, and Au+ zeolite materials and add only a few remarks concerning other d10-zeolites, e.g. such containing Zn2+, Cd2+, and Hg2+ ions.
During the last few years the physical and chemical properties of Cu+–zeolites have attracted some interest in the field of catalysis and of physical adsorption or chemisorption of small molecules. For example Cu+–Y can be applied not only as a catalyst in organic synthesis,5 but also for the conversion of NOx into N2 and O2,6 and for separation procedures like diene/olefin separation.7 Ag+–Y was tested for the same separation procedures,8 but it exhibited lower separation factors. Ag+-containing zeolites were found to oxidize water in a photochemical reaction,9,10 however, the role of the zeolite in these experiments remained obscure. Ag(CN)2−/Au(CN)2− doped zeolite A was studied as a catalyst for the photochemical decomposition of carbaryl pesticide.11 The understanding of the electronic structure of Ag+–A,12,13 recently led to the discovery of the first luminescent silver sulfide quantum dots in zeolite A.14
Knowledge of the coordination sites of metal cations in zeolites is important for the understanding of these materials. They have therefore been studied by X-ray diffraction methods,1,15 by gas adsorption,16 by different kinds of spectroscopy,17 and by theoretical means.18 We show them for the zeolites discussed in this article in Fig. 2, where we have also added SEM pictures to illustrate the morphology of typical materials used in our laboratory.
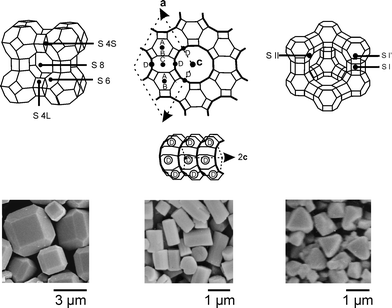 | ||
| Fig. 2 Frameworks of zeolites A, L, and Y showing the crystallographically identified cation positions and SEM pictures of the corresponding zeolite crystals. | ||
The importance of the occupation of a specific site by a cation has been impressively demonstrated for zeolite A, which possesses 8 S6, 3 S8, 6 S4L, and 12 S4S sites. For Ag+–zeolite A it was proven that only if a silver ion sits on one of the S4L positions, does the zeolite turn to deep yellow under evacuation at room temperature,12,13 a reversible color change that was first reported by Rálek et al. in 1962,19 but was misinterpreted for many years. A classical experiment concerns the occupancy of site S8 by means of argon adsorption experiments, which give access to the pore diameter and the inner volume.1,20 We show the result of an Ar adsorption experiment at 87 K for a Ca6A-, a Ag12A-, and a Na12A zeolite in Fig. 3. Ca6A shows a Langmuir type isotherm, so called type I isotherm. The inner volume of Na12A and Ag12A is, however, not accessible for Ar, because the cation positioned at the S8 ring blocks the entrance of the α-cage.
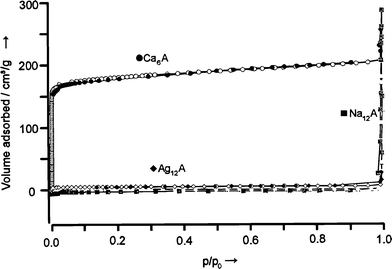 | ||
| Fig. 3 Argon adsorption isotherm of Ca6A-, Ag12A-, and Na12A-zeolites, measured at the boiling point of Ar (87 K). p/p0 is the relative pressure; p0 is the saturation pressure of Ar. | ||
2 Preparation of d10-zeolite materials
A variety of procedures have been used to prepare d10-zeolite materials. One procedure is ion exchange in some cases followed by selective reduction or other selective reactions. An example is the ion exchange with Cu2+ from an aqueous solution, followed by selective reduction with H2 or CO.21 A different procedure is the insertion of molecules or ion pairs from concentrated solutions. In this way anions can be occluded which otherwise would not enter the negatively charged framework. Examples are the inclusion of Cl−, Br−, I−,22 and CN−23 in zeolite A from boiling saturated aqueous solutions of e.g. the corresponding Na+ or Ca2+ salts, followed by e.g. Ag+ exchange. Ag(CN)2− and Au(CN)2− loaded zeolite A was prepared by reaction of potassium zeolite A for 12 h at room temperature with 4 M aqueous KAg(CN)2 and KAu(CN)2, respectively.11 Another procedure is insertion of molecules from the gas phase into a dried zeolite, sometimes followed by a specific reaction. Examples are the insertion of AuCl3 followed by reduction with CO24 and the insertion of CuCl.25Care has to be taken in any case, that the procedure does not damage the zeolite framework. X-Ray powder diffraction data and the morphology should be checked. A good example is the exchange of Cu2+ which works well for zeolites Y and X but which for zeolite A can only be used for low partial exchange, namely 1% to 2.5% by charge, depending on the co-cations. The zeolite framework breaks down at higher Cu2+ exchange as can first be seen by the corrugation of the crystal surface in SEM or TEM pictures, and at greater damage also in X-ray diffraction. It is reasonable to assume that strain is induced in the zeolite framework upon exchange of alkali cations by the Cu2+. The cause of this strain is not the charge—materials which are fully exchanged with Ca2+ are perfectly stable—but the specific coordination geometry. This strain increases with increasing exchange level until the framework ruptures.26 This problem can be avoided by ion exchange of Cu+ in liquid ammonia21 or by insertion of CuCl from the gas phase.27 In this case nearly a total exchange has been obtained without destroying the zeolite framework.
A general procedure for the preparation of quantum sized Ag2S,14 CdS,28 and ZnS28 clusters in zeolites involves first an ion exchange with Ag+, Cd2+, or Zn2+, then drying in vacuum and reacting with H2S. For the preparation of ZnO and CdO nanoparticles, the ion exchange is followed by a calcination procedure.29
Ag+ loaded zeolites can be easily prepared by ion exchange usually from an aqueous silver nitrate or silver perchlorate solution. This ion exchange is fast and can be quantitatively controlled. The equilibrium and also the kinetics depend of course on the type of cations to be exchanged. It is especially easy to see this for sodium or potassium cations. The experimental procedure for ion exchange in the older literature often recommends exchange times of several days, which is not necessary and which can even damage the morphology of the crystals. We illustrate this by means of a simple kinetics experiment of an Ag+ ion exchange at 20 °C in Fig. 4. It shows that ion exchange was complete in less than 20 min.
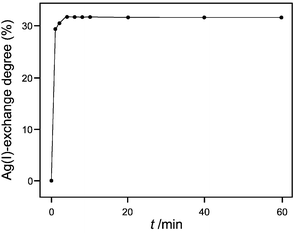 | ||
| Fig. 4 Ag+-exchange degree of zeolite A as a function of the exchange time at 20 °C. A 9.1 mg sample of zeolite A was added to 200 mL of an aqueous 1 M NaNO3 + 1 mM AgNO3 solution. The Ag+ content of the aqueous phase was measured by means of a Ag+ ion selective electrode.30 | ||
The ion exchange isotherm of the system Na+/Ag+–A in Fig. 5 illustrates the strong preference for Ag+ over Na+. We observe a fast quantitative uptake up to an exchange degree of about 0.5 (that means for zeolite A 6 Ag+ per α-cage). The system Ca2+/Ag+–A shows quite a different isotherm. Because of the stronger bonding of the Ca2+ ions to the zeolite framework, the Ag+ ions are less preferred than in the Na+/Ag+–A system and also the exchange rate is slower. It takes about 1.5 h for the equilibrium to be reached. The exchange isotherm of the system Na+/Ag+–ZK4 rises less steeply than the isotherm of Na+/Ag+–A, but Ag+ is still preferred over Na+.
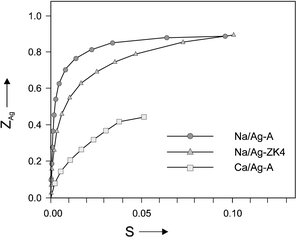 | ||
| Fig. 5 Ion exchange isotherms for the systems Na+/Ag+–A, Na+/Ag+–ZK4 and Ca2+/Ag+–A at room temperature, where ZAg is the exchange degree inside the zeolite and S is the equivalent fraction of Ag+ in solution.13 | ||
3 The electronic structure of d10- zeolites
The simplest approach, which has proven to be very successful, is to regard the electronic structure of a typical zeolite material to a first approximation as a superposition of the electronic structure of the framework, of the charge compensating cations, of the present solvent molecules (usually water), and of the guest species. We start with the band structure and density of states of the silicon dioxide analogue of zeolite A, which is especially simple because of the absence of co-cations. The result of a band structure calculation of a silicon dioxide analogue of zeolite A displayed in Fig. 6 shows that the bands in the HOMO region are flat which indicates the presence of nonbonding states. Some bands below −14 eV are significantly bent and contribute to the Si–O bonding. Further insight is gained from the density of states DOS(E), defined in such a way that DOS(E)dE is the number of states in the interval E to E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +dE. Since we are expressing the crystal orbitals as a linear combination of atomic orbitals (LCAO) we can highlight specific atomic orbitals or linear combinations of them. This is done by shading the oxygen 2p contributions and leaving the 2s oxygen and the silicon contributions blank. This shows that the HOMO region consists of nearly pure oxygen 2p lone pairs which we denote as |O<. The DOS of the LUMO region reflects the loose framework of the material.
+dE. Since we are expressing the crystal orbitals as a linear combination of atomic orbitals (LCAO) we can highlight specific atomic orbitals or linear combinations of them. This is done by shading the oxygen 2p contributions and leaving the 2s oxygen and the silicon contributions blank. This shows that the HOMO region consists of nearly pure oxygen 2p lone pairs which we denote as |O<. The DOS of the LUMO region reflects the loose framework of the material.
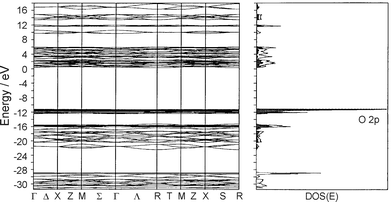 | ||
| Fig. 6 Band structure (left) and density of states (right) of the silicon dioxide analogue of zeolite A. The oxygen 2p density is projected out (shaded regions). | ||
The Al3+ centers in zeolites cause a negatively charged framework (AlO2−). This charge is compensated by exchangeable cations, which influence the band structure to some extent. Calculations show that the HOMO and the LUMO regions are in general similar to those illustrated in Fig. 6. Next we add a solvent. We only discuss water as a solvent molecule. The only level of importance is its lone pair at −12.6 eV, the energy of which is little affected e.g. by coordination to silver cations.31 We can therefore assume that the HOMO–LUMO region of zeolites containing Cu+, Ag+, and Au+ can be drawn qualitatively as illustrated in Fig. 7. We show on the left a correlation diagram and on the right a simplified state diagram. It also holds for the alkali ions Li+,...,Cs+, but in this case the d-levels can be omitted. The levels consist of the oxygen lone pair region denoted as |O<, the empty ns′ level of the metal cations M+, and of the LUMO region of the zeolite which may be modified by np′ contributions of M+. The ns′ and np′ and also the (n−1)d levels are shifted to higher energy with respect to the ns and np levels of the free cations because of antibonding interactions with the oxygen lone pairs. This scheme strongly suggests the occurrence of ligand to metal charge transfer (LMCT) transitions of the ns′←|O< type, where an oxygen lone pair electron is excited to the metal cation coordinated to the zeolite oxygen. The energy ΔECT needed for this transition is equal to the difference of the ionization potential Ip|O< of the oxygen lone pair and the first ionization potential IpM of the metal M, plus a correction ΔM which stands for the antibonding interaction of the empty ns′ level of the metal ion M+ with the environment and maybe some shift of the potential energy curve. The index M of the interaction term ΔM indicates that the destabilization of the ns levels depend on the nature of the cation. We shall see in the next section, that it also depends on the coordination site, a fact we ignore at the moment.
| ΔECT( ns′←|O<) = Ip|O< − IpM + ΔM | (1) |
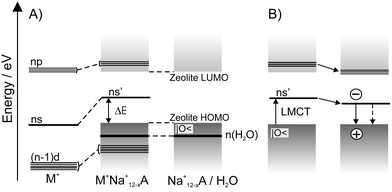 | ||
| Fig. 7 A) Correlation diagram of M+(d10) loaded zeolite. On the left the energy levels of M+, on the right the energy levels of a zeolite are shown. The HOMO region of zeolites consists of many narrow lying localized states strongly concentrated on the oxygen atoms. We call this the lone pair region of the silicate and abbreviate it as |O<. The water lone pairs are additionally marked. B) Energy diagram of metal cations in a zeolite framework. Some of the np′ levels may reach into the LUMO region of the silicate. Three lines have been added to indicate this. | ||
This simple relation allows us to estimate the energy of the charge transfer band for different situations. ns′←|O< charge transfer transitions have been observed in Cu+–A, Cu+–X and Ag+–A zeolites in the region of 28000 cm−1 (360 nm). This means that ΔM is in the order of 4000 cm−1 for both metals, because the first ionization potentials of the Cu and Ag atoms are 7.68 and 7.54 eV, respectively. CT transitions for which ΔM is larger will be discussed in the next section. Ag+ in water absorbs light at about 225 nm,31 in agreement with eqn. 1. Au has the largest IpM, namely 9.18 eV, which gives Au its noble character. In general Au+ is unstable but can be stabilized by specific ligands. This makes it difficult to incorporate Au+ by conventional ion-exchange methods. However, some researchers have reported the synthesis of Au+ loaded zeolites by means of ion exchange with gold complexes or by sublimation of (AuCl3)2.32,33 The electronic spectra of the resulting materials so far reported suggested that Au+ was still coordinated to chloride and direct interaction to the zeolite could not be observed. Clear assignment of the observed electronic transitions have not been established. More work is needed to get electronic spectra of Au+ coordinated to zeolite oxygen atoms.
Luminescence of the Cu+–A, Cu+–X, Cu+–ZSM5, and Ag+–A zeolites after ns′←|O< excitation occurs at 400–700 nm, depending on the samples and the conditions.25–27 This means that the Stokes shift is in the order of 8000 cm−1. Excitation of an electron from the oxygen lone pair level |O< into the empty ns′ orbital of the metal cation causes a formal reduction of M+ to M0. The radius r of M+ is significantly smaller than that of M0 (r(Cu+) = 0.96 Å, r(Cu0) = 1.35 Å, r(Ag+) = 1.26 Å, r(Ag0) = 1.6 Å). This means that the LMCT transition expands the metal by 0.3–0.4 Å which causes a change in its position. As a consequence, the ns′ level relaxes to a state of lower energy which we denote as the (|O<)⊕(ns′)1 state. The latter relaxes to the ground state either by emitting a photon with a large Stokes shift or by radiationless processes, as illustrated in Fig. 7 on the right hand side.
The luminescence intensity of Cu+ and Ag+ zeolites is remarkably influenced by the degree of hydration of the samples. Fig. 8 shows the luminescence spectra of Cu9.3H9.3Na67.4X as a function of the degree of hydration. This sample was prepared by a partial ion exchange of Na+–X with an aqueous CuCl2 solution at room temperature and selective reduction of Cu2+ to Cu+ with CO.26 At a water vapor pressure of 16 mbar the sample can be regarded as fully hydrated. Maximum emission is achieved at a well defined range of water content. While in the case of Cu+–A and Cu+–X the emission maximum only shifts 10 nm during rehydration, the luminescence of Ag+–A shows more dynamics. In both cases, however, the optimum amount of water depends on the co-cations and on the type of zeolite. For example, while Ag+/Na+–A doesn’t show luminescence in its fully hydrated state, a weak luminescence can be observed in Ag+/Ca2+–A and Ag+/Mg2+–ZK4. An explanation for this phenomenon can be attempted by considering the stronger electrostatic interaction between divalent cations and water than between monovalent cations and water. Thus in zeolites with divalent co-cations like Ca2+ and Mg2+, less water is coordinated to Ag+ than in e.g. Ag+/Na+–A. There are not many reports about luminescence of Au+–zeolites. However, luminescence of Au(CN)2− loaded zeolite A occurs at 390–445 nm and is assigned to trimers and tetramers of the complex. This also suggests that the Au+ ions do not have strong interactions with the zeolite.
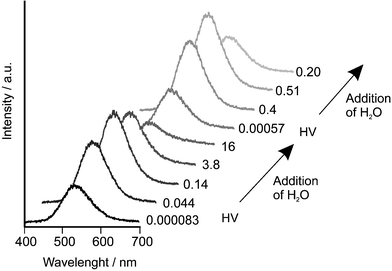 | ||
| Fig. 8 Luminescence spectra of Cu9.3H9.3Na67.4X as a function of the degree of hydration measured at room temperature after excitation at 355 nm. Two evacuation–hydration cycles are shown. The numbers on the right side of the spectra are the pressure in the sample compartment in mbar. HV means 30 min of evacuation at room temperature.26 | ||
4 The electronic structure of Ag+ zeolite A
Rálek et al. reported in 1962 that hydrated colorless zeolite Ag+xNa+12−xA turns yellow to brick-red on activation.19 No explanation of this phenomenon was given at that time. Later it was believed that the color change was due to formation of silver clusters (Ag0n) in the cavities of silver zeolite A. These neutral silver species were assumed to be formed at elevated temperatures via an auto-reduction process in which O2 from the zeolite framework was released.34 We studied the vibrational spectra of Ag+ zeolite A materials in some detail,35 and we showed that activation at room temperature under high vacuum is already sufficient to produce the yellow form of Ag+xNa+12−xA. The fully reversible color change, which depends on the hydration state of the silver zeolite, was attributed to electronic charge transfer transitions from the oxygen lone pairs of the zeolite framework to the empty 5s orbital of the Ag+ ions, denoted as Ag+(5s)←O(n).12 Silver-containing sodium zeolite A is colorless in its fully hydrated form. In activated silver zeolite A materials, the Ag+ is forced to coordinate zeolite oxygen because an insufficient number of available water molecules are present. The question remained if specific coordination sites which act as yellow and/or red ‘color centers’ can be identified. We answered this question by studying the UV/vis spectra of Ag+xNa+12−xA and of Ag+xCa2+6−0.5xA materials in their fully hydrated, in HV room temperature dehydrated, and in HV elevated temperature dehydrated states. The possible ion positions in Ag+xCa2+6−0.5xA, probed by gas adsorption experiments, offered the unique possibility of investigating different coordination sites of Ag+ ions in zeolite A.13 Pure sodium and calcium zeolite A do not absorb light within the spectral range from 200–1000 nm we have investigated. This means that any absorption band or colors observed in silver zeolite A materials are due to the presence of silver ions. Fig. 9 shows the different colors of a Ag+6Na+6A zeolite in its fully hydrated state (white), after activation under high vacuum (2 × 10−7 mbar) at room temperature (deep yellow) and after heating in the vacuum to 200 °C (red). The spectrum of the yellow sample is fully reversible. After readsorption of water the yellow samples turn white again and the absorption spectrum is the same as before the activation.
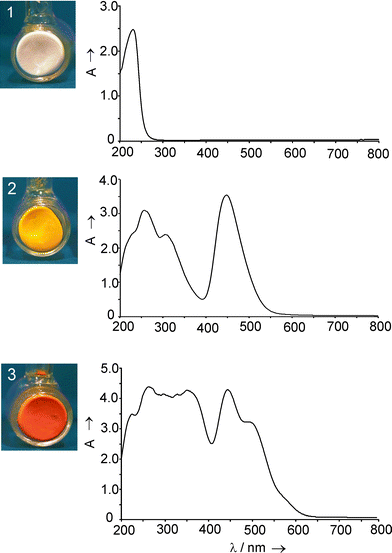 | ||
| Fig. 9 Samples (left) and the corresponding UV/Vis spectra (right) of Ag+6Na+6A in its fully hydrated state (1), after activation in high vacuum at room temperature (2), and after activation in high vacuum at 200 °C (3). | ||
We found that 6- and 8-ring coordinated Ag+ give rise to electronic transitions in the near UV region. An absorption in the visible, namely at 450 nm, was only observed in materials where 4-ring coordinated Ag+ was present and only they showed the typical deep yellow color. We also observed that Ag+ avoids the 4-ring sites as long as possible in Ag+xCa2+6−0.5xA, namely as long as x is smaller than 10. In the case of Ag+xNa+12−xA either a Na+ or a Ag+ is forced to coordinate a 4-ring site because all other places are occupied. The presence of the 450 nm absorption responsible for the yellow color, already at x < 0.2, proves that isolated Ag+ ions are sufficient to cause it and that the 4-ring coordination of Ag+ is significantly stronger than that of the Na+. The red color of elevated temperature activated samples is caused by a strong absorption band at 520 nm. We observed that samples which remained colorless after room temperature activation never turned red, that samples with lower silver content than one Ag+ per α-cage never turned red, and that room temperature dehydration under our experimental conditions was not sufficient to produce red colored samples. These observations strongly indicate that only samples with 4-ring coordinated Ag+ can give rise to the 520 nm band and this only occurs if a second Ag+ is not too far away at a 6-ring site, so that they can interact to develop a corresponding low lying state.
Molecular orbital calculations carried out on a sufficiently large zeolite part consisting of 1296 atoms allowed us to address questions about the nature of the HOMO and of the LUMO region, about the contributions of the zeolite framework atoms to the electronic transitions, about the influence of the local symmetry of the Ag+ at 4- and at 6-ring sites, and about the importance of Ag+–Ag+ interactions. We found that 6-ring coordinated Ag+ give rise to electronic transitions in the near UV and that the 4-ring coordinated Ag+ is responsible for the deep yellow color of the room temperature activated material. This implies that similar Ag+(5s) ← O(n) LMCT transitions are to be expected in other Ag+ exchanged zeolites. Ag+ exchanged zeolite Y can be used as a test. We therefore measured UV/vis spectra of pure Na+69Y, of room temperature HV dehydrated Ag+69Y, and rehydrated Ag+69Y. The main result is that an intense band at about 34000 cm−1 appears upon dehydration which vanishes upon rehydration.13 Analogously one would expect a similar type of LMCT transitions in Cu+ zeolite materials. Cu+(4s)←O(n) LMCT transition, reversible upon HV hydration/dehydration, have indeed been observed in Cu+ zeolite A and X.26
Based on all the information which has been collected over the last few years, we can now draw the schematic state diagram in Fig. 10 for Ag+ containing zeolites.
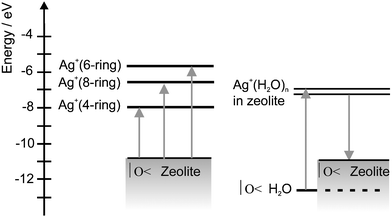 | ||
| Fig. 10 State diagram of Ag+-loaded zeolite A. On the left side we show the levels observed in room temperature activated zeolites in which all three sites are occupied by silver ions, while the scheme on the right side corresponds to situations typically observed in Ag+xCa2+6−0.5xA materials containing some water. | ||
5 Quantum dots embedded in zeolites
The well defined cavities and channels of zeolites provide a convenient environment for preparing clusters and cluster arrays with a narrow size distribution as discussed in a review by G. Stucky and Mac Dougall.36 d10-Metal ion clusters of ZnO, ZnS, CdO, CdS,28,29,37,38 of silver halides22,39 and more recently of silver sulfide14 were reported. The preparation of the latter was based on the observation discussed in Section 4 that Ag+ loaded zeolites can be reversibly activated at room temperature which allows the application of very mild synthesis conditions. This synthesis principle is expected to be applicable for the preparation of many other zeolite based host–guest materials. We therefore focus on the description of this most recent material.α-Ag2S is a semiconductor with a monoclinic structure and a band gap of approximately 1 eV at room temperature. Fig. 11 on the left shows the calculated density of states (DOS) of bulk α-Ag2S. The electronic transition from valence band to conduction band is essentially a charge transfer from 3p(S) to 5s(Ag). This property is also observed in a Ag2S molecule. The HOMO–LUMO region of such a molecule, and presumably also of larger silver sulfide clusters, fits well into the badgap of zeolite A (see right side of Fig. 11), therefore giving rise to a variety of electronic transitions.
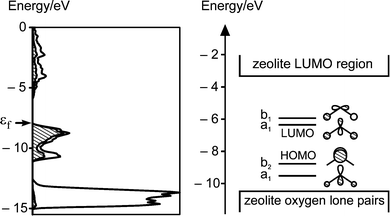 | ||
| Fig. 11 Left: Density of states (DOS) plot of α-Ag2S. The hatched region indicates the contribution of sulfur 3p-states. The Fermi level εf is marked by an arrow. Right: HOMO–LUMO region of a Ag2S molecule in comparison to the HOMO–LUMO region of zeolite A. | ||
Bulk silver sulfide has been considered for photoimaging and photodetection in the IR,40 while small clusters are known to play an important role in photographic sensitivity.41,42 It has been reported that silver sulfide clusters with sizes ranging from 23 Å to 76 Å can be synthesized in reverse micelles. Another method utilizes the rapid expansion of a AgNO3 solution in supercritical ammonia into an ethanol solution of Na2S. An average diameter of 73 Å was found for particles formed in this way after stabilizing with a suitable polymer. Dosed addition of a AgNO3 solution to a gelatin solution containing Na2S was reported to yield silver sulfide clusters in the size regime between 30 Å and 100 Å. Other methods use thin nylon films, Nafion membranes or capping with cysteine/glutathione to stabilize the silver sulfide clusters.43 Some of the results obtained by the above mentioned methods suggest the presence of a quantum size effect for silver sulfide clusters with a diameter between 20 Å and 100 Å while other reports clearly negate the presence of such an effect; for references see ref. 14.
Quantum sized silver sulfide clusters have been synthesized in the cavities of sodium and calcium zeolite A and ZK-4 microcrystals by exposing the room temperature activated Ag+-loaded zeolite to H2S. The formation of Ag2S was also detected by X-ray photoemission spectroscopy (XPS) in zeolite Y after exposure of Ag+–Y to H2S.8 The growth of the silver sulfide clusters in zeolite A and ZK-4 during rehydration of the samples was investigated by means of diffuse reflectance spectroscopy. Monomers of Ag2S are formed at low Ag+-loading through reaction of AgSH molecules, yielding colorless composites, which exhibit a characteristic blue-green photoluminescence. Yellow colored samples showing an orange-red luminescence with an average decay time of 81 μs (−160 °C) are obtained at medium silver sulfide content. Further increasing the silver sulfide loading and therefore the cluster size causes a bathochromic shift of this emission which is accompanied by a shortening of the luminescence lifetime. The samples generally exhibit large Stokes shifts, which can be attributed to HOMO–LUMO transitions with small oscillator strengths. The Ag2S–zeolite host–guest system constitutes a three-dimensional array of silver sulfide clusters. The experimental data and quantum chemical calculations show that the optical absorption and luminescence properties of this material are mainly due to the presence of isolated silver sulfide clusters inside the zeolite cavities. The characteristics of the composites are thereby to a certain extent influenced by the co-cations. Most remarkably, stronger luminescence, easily visible at room temperature, could be observed for silver sulfide clusters in calcium zeolite A as compared to clusters in sodium zeolite A. We show some recent observation on Ag2S–CaA-x materials to illustrate this: x denotes the average number of silver atoms per pseudo unit cell. Samples with low silver loading (e.g. x = 0.01 Ag+) are white but show green luminescence. Fig. 12 displays as an example the spectra of Ag2S–CaA-0.01.
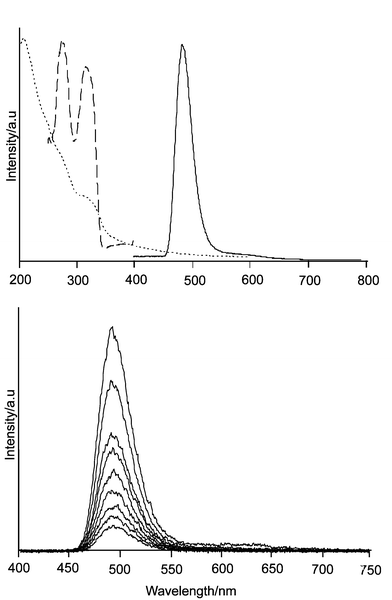 | ||
| Fig. 12 Top: Emission (solid), excitation (dashed), and diffuse reflectance (points) spectra of Ag2S–CaA-0.01. Luminescence was observed after 320 nm excitation while the excitation spectrum was measured at the maximum of the emission which is at 490 nm, both at −190 °C. The diffuse reflectance spectrum (Kubelka–Munk) was measured at room temperature. Bottom: Luminescence spectrum of the same sample after excitation at 320 nm, observed at different times after excitation (time window 100 μs). Mono exponential decay was observed with a luminescence lifetime of 300 μs. | ||
Analysis of our data shows that these are the spectra of isolated Ag2S molecules (quantum dots) in the three dimensional framework of zeolite A. This is the first time that the spectrum of an isolated Ag2S molecule has been observed; see also Fig. 4 of ref. 14. A second luminescence band is observed when the silver loading is increased. Samples in the loading range x > 0.1 up to x = 1 show deepening of the yellow color. They exhibit both luminescence bands: the 490 nm and the new 620 nm emission; see Fig. 13. With increasing silver sulfide loading the 620 nm emission gets more intense and the 490 nm emission disappears. This means that the ratio of the amount of Ag2S molecules and larger silver sulfide clusters in the zeolite can be tuned by choosing different initial amounts of Ag+. The two bands show a different intensity dependence on the temperature. This means that e.g. an x = 1 material could be used as a responsive micro temperature sensor, by comparing the intensity ratio of the two bands.
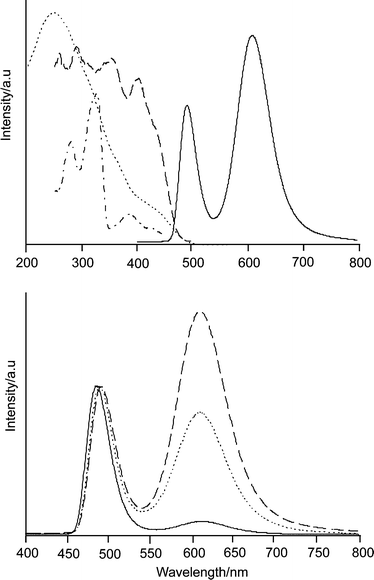 | ||
| Fig. 13 Top: Ag2S–CaA-1: luminescence (solid) and excitation spectra (dashed observed at 620 nm, dash dot observed at 490 nm) at −190 °C, and diffuse reflectance spectrum (Kubelka–Munk) at room temperature. Bottom: Luminescence spectra of Ag2S–CaA-x with x = 0.1 (solid), x = 0.75 (dot), and x = 1 (dashed) after excitation at 320 nm. The spectra are scaled to the same height of the short wavelength band. | ||
The luminescence spectrum in Fig. 13 can be broken down into two components. The 620 nm emission can be specifically excited at λ > 400 nm. It has a decay time of about 100 μs. This is in contrast to the 490 nm band which decays much slower. The 490 nm band corresponds in every respect to the luminescence shown in Fig. 12 which means that it stems from Ag2S dots. We have good reasons to assign the 620 nm emission to (Ag2S)2 clusters and we are now investigating if and to what extent radiationless excitation energy transfer from excited Ag2S to (Ag2S)2 cluster does occur. Similar results were observed by using zeolite ZK4 as a host, instead of zeolite A. Increasing loading shifts both the absorption and the emission bands to longer wavelengths, which is most probably due to the formation of larger clusters. The synthesis conditions and the material’s properties are influenced by the co-cations (Li+, Na+, K+, Mg2+, Ca2+, Sr2+), which is not astonishing in principle. However, the reasons for this are not well understood and more systematic work is needed. This is possible because the results are reproducible, which means that the system can be controlled.
We summarize that the reaction of activated Ag+-loaded zeolite A with H2S leads to the formation of silver sulfide clusters in the zeolite cavities. The size of the clusters can be controlled by adjusting the initial Ag+-loading. Rehydration of the reaction products contributes to the cluster growth by enhancing the mobility of small silver sulfide and silver hydrosulfide species. The reaction between AgSH molecules, which are formed at low Ag+-loading levels, produces Ag2S and H2S. The formation of Ag2S monomers and also of larger silver sulfide clusters is discernible by a band above 300 nm in the diffuse reflectance spectra of the composites. Considering the dimensions of an α-cage of zeolite A, the largest silver sulfide cluster that can be synthesized in such a cavity would have a stoichiometry of approximately Ag8S4. Monomers of Ag2S have to be considered as the smallest clusters. These species are responsible for the blue-green luminescence exhibited by samples of low silver sulfide content. At higher silver sulfide content a luminescence band around 620 nm is observed. The red-shift of this band with increasing silver sulfide loading can be attributed to a quantum size effect. The comparatively long average luminescence lifetimes observed for the orange-red emission in Ag2S–CaA (e.g. 81 μs for Ag2S–CaA-2 at −160 °C) and the large Stokes shifts are in agreement with the results of the MO calculations, which yield small oscillator strengths for the HOMO–LUMO transitions, but very large oscillator strengths for transitions of higher energy. According to these results, the qualitative state diagram shown in Fig. 14 can be constructed. Because of the inversely proportional relation between the oscillator strength of an electronic transition and the corresponding intrinsic emissive lifetime, the intense absorption transitions give rise to short-lived excited states. Non-radiative relaxation into the lowest excited state is followed by the observed long-lived luminescence. The Ag2S–zeolite host–guest systems constitute three-dimensional quantum dot arrays or quantum lattices. In the case of Ag4S2 clusters, coupling over the whole lattice is expected to occur at an average intercluster distance below 10 Å (center to center).14
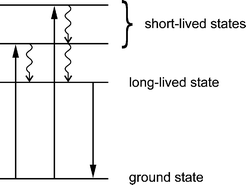 | ||
| Fig. 14 State diagram for small luminescent silver sulfide clusters. This is valid for Ag2S and Ag4S2. | ||
6 Photocatalytic water oxidation
The splitting of water into hydrogen and oxygen driven by solar energy is considered as the ‘Holy Grail’ towards a clean and renewable energy source.44 One of the key problems in photocatalytic water splitting has always been the oxidation of water to oxygen driven by visible light, mainly because of the difficulty in accumulating the four oxidative equivalents required. For several years it was known that aqueous dispersions of Ag+ zeolites are able to produce oxygen under illumination, however, the conditions were not clearly defined. We observed that self-sensitization of photo-oxygen evolution occurs in this system. This means that dispersions which were initially insensitive to visible light become photoactive after they have been illuminated for a few minutes with near UV light.10 This system, however, was not catalytic and the role of the zeolite was not understood. We later found that aqueous dispersions of very pure Ag+ exchanged zeolites were not photoactive, but that zeolites containing silver chloride were strongly photoactive provided that an excess of Ag+ was present. Oxygen evolution was observed in a pH range of about 4 to 8. In fact the zeolite was found not to be necessary for the photochemical oxygen evolution from silver chloride. But the system was still not photocatalytic. Fortunately we found means to prepare silver chloride based electrodes in which the AgCl acts as a catalyst in the presence of a small excess of silver cations with a maximum oxygen evolution rate between pH 4 and 6. Its photoactivity extends from the UV to the visible region due to a process known as self-sensitization. The photocatalytic reaction consists of two main processes: water is oxidized to oxygen plus protons, and silver cations are reduced to silver upon irradiation. The reduced silver species are reoxidized which makes the system catalytic.45,46 The self-sensitization of the photochemical activity of AgCl is due to silver clusters adsorbed on its surface. A comparison of the energetic positions of the AgCl band gap47 and the silver clusters shows that the silver clusters have empty energy levels below the conduction band of AgCl (see Fig. 15).48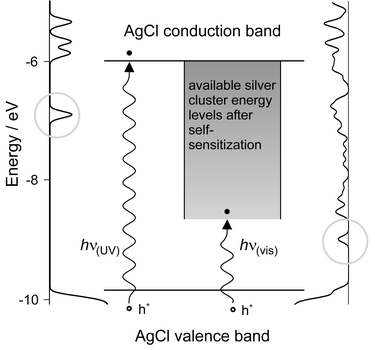 | ||
| Fig. 15 Mechanism of self-sensitization. The density of states (DOS) of a silver chloride cluster is shown on the left and the local DOS of silver–silver chloride cluster composite is shown on the right. We observe new induced levels below the LUMO of the AgCl cluster. A schematic view is shown in the middle. The lowest lying levels into which electrons can be excited are marked with a circle (left: surface states, right: metal induced gap states). The position of the valence band is not markedly influenced by the presence of the silver cluster.48 | ||
In the absence of silver clusters, AgCl does not absorb light below the indirect band gap transition, which is in the near UV at 3.3 eV (380 nm). Their presence enables a new electronic transition from the AgCl valence band to empty silver cluster energy levels. The energy for this transition is lower than the energy needed for an optical transition from the AgCl valence band to the AgCl conduction band. Nevertheless, this new optical transition in the visible spectral range still initiates the oxidation of water, because the conduction band is not directly involved in water oxidation.31 Thus, the photocatalytic oxidation of water on AgCl is extended from the near UV into the visible range of the spectrum. Self-sensitization as observed by us should not be confused with the spectral sensitization effected by silver clusters, also termed photographic Becquerel effect. The latter observation has been attributed to an electron injection from electron-donating silver clusters into the AgCl conduction band. The first investigation of the photo-electrochemical properties of AgCl-coated electrodes was made by Becquerel in 1839,49 a few years after Talbot had realized the first photographic pictures on paper based on AgCl.50 The photo current observed by Becquerel was reductive.
Our experimental results show, that appropriately prepared silver chloride electrodes can fulfil this task and we therefore consider this material as a suitable choice in a photocatalytic water splitting device.31,45,46 However, the sensitivity of the silver chloride based photon anode is not satisfactory. We know that it can be improved, if the electrode is first covered with a monolayer of a Ag+ exchanged zeolite A as described in ref. 30 and then modified with a thin layer of AgCl. An experiment which shows the photo response of such an electrode is shown in Fig. 16. The experiment was carried out under similar conditions as reported in ref. 45. The AgCl modified zeolite layer has a maximum O2 production rate that is almost 4 times that of a AgCl layer on a gold modified conducting glass electrode which is likely due to its increased active surface area. However, the O2 production rate is not sustained and falls to almost half its initial value by the end of the first cycle. One reason for the abrupt fall is the lack of electrical contact between the reduced silver on the surface of the zeolite (a product of the photo-reaction) and the conducting substrate. Without reoxidation of these reduced silver species the amount of photoactive AgCl on the surface cannot be maintained, hence the drop in O2 production rate. That notwithstanding, the results show that there is much possibility for improvement by modifying the AgCl layer structure. Impressive progress on the preparation of zeolite monolayers has been made over the last few years, which should make it possible to combine the more reactive AgCl modified zeolite electrode with the stability and reversibility of a AgCl layer on a gold modified electrode.
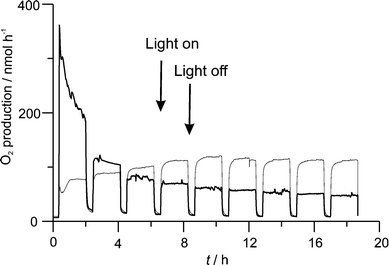 | ||
| Fig. 16 Photo electrochemical experiment, illustrating the difference of the oxidation of water to O2 for a AgCl surface modified zeolite-A electrode (bold), and for a AgCl layer on a gold modified conducting glass electrode. The corresponding current, about 8 μA during light period for the layer on gold, 0 for the zeolite layer, is not shown; see refs. 45 and 46. | ||
7 Concluding remarks
A variety of procedures have been used to prepare d10-zeolite composites. The electronic structure of these materials can be regarded to a first approximation as a superposition of the framework, of the charge compensating ions, of solvent molecules and of guest species. Zeolite oxygen to d10-ion charge transfer transitions dominate the electronic spectra if the ions coordinate to the zeolite oxygens. Specific coordination sites can influence the energy and the intensity of these transitions remarkably. The reversible color change from colorless to deep yellow of Ag+–A and Ag+–ZK4 materials, which is very specific for Ag+ ions coordinated to the 4-ring, is a beautiful demonstration of this fact. Intra guest transitions dominate in quantum dot materials. We discuss this in detail for luminescent Ag2S zeolite A. The possibility of preparing highly organized quantum dot materials opens a challenging world of possibilities in the assembly of highly organized macroscopic structures. We have discussed that the zeolite is not needed for the photocatalytic water oxidation on Ag+/AgCl photoanodes with visible light but that it can be used to increase the active surface area substantially. The challenge of developing reversible Ag+/AgCl photoanodes for water oxidation to di-oxygen based on this fact remains to be explored.8 Acknowledgements
We acknowledge financial support by the Schweizerisches Bundesamt für Energiewirtschaft (project no. 36846) and the Schweizerische Nationalfonds (project no. 20-53414.98).9 References
- D. W. Breck, Zeolite Molecular Sieves, John Wiley, New York, 1974 Search PubMed.
- D. L. Griscom, J. Non-Cryst. Solids, 1977, 24, 155–234 Search PubMed.
- G. Calzaferri and R. Hoffmann, J. Chem. Soc., Dalton Trans., 1991, 917–928 RSC.
- T. Jüstel, D. U. Wiechert, C. Lau, D. Sendor and U. Kynast, Adv. Funct. Mater., 2001, 11, 105–110 CrossRef CAS.
- S. T. King, J. Catal., 1996, 161, 530–538 CrossRef CAS.
- M. Anpo, M. Matsuoka, K. Hanou, H. Mishima, H. Yamashita and H. H. Patterson, Coord. Chem. Rev., 1998, 171, 175–184 CrossRef CAS.
- A. Takahashi and R. T. Yang, Langmuir, 2001, 17, 8405–8413 CrossRef CAS.
- A. Takahashi, R. T. Yang, C. L. Munson and D. Chinn, Ind. Eng. Chem. Res., 2001, 40, 3979–3988 CrossRef CAS.
- P. A. Jacobs, J. B. Uytterhoeven and H. K. Beyer, J. Chem. Soc., Chem. Commun., 1977, 128–129 RSC.
- G. Calzaferri, S. Hug, T. Hugentobler and B. Sulzberger, J. Photochem., 1984, 26, 109–118 Search PubMed.
- S. M. Kanan, C. P. Tripp, R. N. Austin and H. H. Patterson, J. Phys. Chem. B, 2001, 105, 9441–9448 CrossRef CAS.
- R. Seifert, A. Kunzmann and G. Calzaferri, Angew. Chem., 1998, 110, 1603 ( Angew. Chem., Int. Ed. , 1998 , 37 , 1521–1524 ) CrossRef.
- R. Seifert, R. Rytz and G. Calzaferri, J. Phys. Chem. A, 2000, 104, 7473–7483 CrossRef CAS.
- D. Brühwiler, C. Leiggener, S. Glaus and G. Calzaferri, J. Phys. Chem. B, 2002, 106, 3770–3777 CrossRef.
- S. B. Jang, U. S. Kim, Y. Kim and K. Seff, J. Phys. Chem., 1994, 98, 3796–3800 CrossRef CAS.
- D. W. Breck, W. G. Eversole, R. M. Milton, T. B. Reed and T. L. Thomas, J. Am. Chem. Soc., 1956, 78, 5963–5977 CrossRef CAS.
- K. H. Lim and C. P. Grey, J. Am. Chem. Soc., 2000, 122, 9768–9780 CrossRef CAS.
- F. M. Higgins, N. H. De Leeuw and Stephen C. Parker, J. Mater. Chem., 2002, 12, 124–131 RSC.
- M. Rálek, P. Jíru, O. Grubner and H. Beyer, Collect. Czech. Chem. Commun., 1962, 27, 142–146 CAS.
- P. E. Hathaway and M. E. Davis, Catal. Lett., 1990, 5, 333–348 Search PubMed.
- D. H. Strome and K. Klier, J. Phys. Chem., 1980, 84, 981–984 CrossRef CAS.
- R. M. Barrer and A. J. Walker, Trans. Faraday Soc., 1964, 60, 171–184 RSC.
- R. J. Taylor, R. S. Drago and J. P. Hage, Inorg. Chem., 1992, 31, 253–258 CrossRef CAS.
- S. Qiu, R. Ohnishi and M. Ichikawa, J. Chem. Soc., Chem. Commun., 1992, 1425–1427 RSC.
- C. Lamberti, S. Bordiga, M. Salvalaggio, G. Spoto, A. Zecchina, F. Geobaldo, G. Vlaic and M. Bellatreccia, J. Phys. Chem B, 1997, 101, 344–360 CrossRef.
- G. Calzaferri, R. Giovanoli, I. Kamber, V. Shklover and R. Nesper, Res. Chem. Intermed., 1993, 19, 31–57 Search PubMed.
- S. Bordiga, G. Turnes Palomino, D. Arduino, C. Lamberti, A. Zecchina and C. Otero Arean, J. Mol. Catal. A: Chem., 1999, 146, 97–106 CrossRef CAS.
- M. M. Garcia, H. Villavicencio, M. Hernandez–Velez, O. Sanchez and J. M. Martinez–Duart, Mater. Sci. Eng., C, 2001, 15, 101–104 Search PubMed.
- M. Wark, G. Schulz-Ekloff and Nils I. Jaeger, Bulg. Chem. Commun., 1998, 30, 129–141 Search PubMed.
- J.-W. Li, K. Pfanner and G. Calzaferri, J. Phys. Chem., 1995, 99, 2119–2126 CrossRef CAS.
- S. Glaus and G. Calzaferri, J. Phys. Chem. B, 1999, 103, 5622–5630 CrossRef CAS.
- Y.-M. Kang and B.-Z. Wan, Catal. Today, 1995, 26, 59–69 CrossRef CAS.
- T. M. Salama, S. Takafumi, R. Ohnishi and M. Ichikawa, J. Phys. Chem., 1996, 100, 3688–3694 CrossRef CAS.
- T. Sun and K. Seff, Chem. Rev., 1994, 94, 857–870 CrossRef CAS.
- J. Baumann, R. Beer, G. Calzaferri and B. Waldeck, J. Phys. Chem., 1989, 93, 2292–2302 CrossRef CAS.
- G. D. Stucky and J. E. Mac Dougall, Science, 1990, 247, 669–677 CrossRef CAS.
- Y. Wang and N. Herron, J. Phys. Chem., 1987, 91, 257–260 CrossRef CAS.
- J. E. Mac Dougall, H. Eckert, G. D. Stucky, N. Herron, Y. Wang, K. Moller, T. Bein and D. Cox, J. Am. Chem. Soc., 1989, 111, 8006–8007 CrossRef CAS.
- A. Stein, G. A. Ozin and G. D. Stucky, J. Am. Chem. Soc., 1992, 114, 8119–8129 CrossRef CAS.
- S. Kitova; J. Eneva; A. Panov and H. Haefke, J. Imaging Sci. Technol., 1994, 38, 484–488 Search PubMed.
- T. Tani, J. Imaging Sci. Technol., 1995, 39, 386–392 Search PubMed; T. Tani, J. Imaging Sci. Technol., 1998, 42, 135–143 Search PubMed.
- R. C. Baetzold, J. Imaging Sci. Technol., 1999, 43, 375–381 Search PubMed.
- M. C. Brelle, J. Z. Zhang, L. Nguyen and R. K. Mehra, J. Phys. Chem. A, 1999, 103, 10194–10201 CrossRef CAS.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- M. Lanz, D. Schürch and G. Calzaferri, J. Photochem. Photobiol. A: Chem., 1999, 120, 105–117 Search PubMed.
- G. Calzaferri, D. Brühwiler, S. Glaus, D. Schürch, A. Currao and C. Leiggener, J. Imaging Sci. Technol., 2001, 45, 331–339 Search PubMed.
- S. Sumi, T. Watanabe, A. Fujishima and K. Honda, Bull. Chem. Soc. Jpn., 1980, 53, 2742–2747 CAS.
- S. Glaus, G. Calzaferri and R. Hoffmann, Chem. Eur. J., 2002, 8, 1786–1794 CrossRef CAS.
- E. Bequerel, C.R. Acad. Sci., 1839, 9, 561 Search PubMed.
- L. J. Schaaf, Out of the Shadows: Herschel, Talbot, and the Invention of Photography, Yale University Press, New Haven, CT, 1992 Search PubMed.
| This journal is © The Royal Society of Chemistry 2003 |