Infrared study of CO and 2-butenal co-adsorption on Zn modified Pt/CeO2–SiO2 catalysts
Joaquin
Silvestre-Albero
b,
A.
Sepúlveda-Escribano
b,
F.
Rodríguez-Reinoso
b and
James A.
Anderson
*a
aCatalysis and Surface Chemistry group, Division of Physical and Inorganic Chemistry, The University, Dundee, Scotland, UK DD1 4HN
bDepartamento de Química Inorgánica, Universidad de Alicante, Aptdo. 99, E-03080 Alicante, Spain
First published on 26th November 2002
Abstract
Infrared spectra of CO alone and CO followed by the introduction of 2-butenal (crotonaldehyde) have been recorded for Pt/CeO2–SiO2 and Zn doped Pt/CeO2–SiO2 catalysts which had been pre-reduced at 473, 623 and 773 K. Samples treated at the highest reduction temperature showed the greatest selectivity toward the unsaturated alcohol although no clear spectroscopic evidence for the formation of an SMSI state was obtained. Although 773 K reduced Zn containing catalysts showed the greatest yield, there was no IR spectroscopic evidence to support the formation of significant quantities of a Pt–Zn alloyed phase or that the addition of Zn enhanced Pt-support interactions beyond increasing the amount of surface reduced ceria. Results show that, contrary to popular opinion, the development of an SMSI state which enhances the density of support-metal interface sites is not essential in order to increase the yield of unsaturated alcohol and the presence of reduced cationic sites at the interface zone may be the crucial factor. Although IR spectra in the νCO region were dominated by dipole coupling effects, enhanced intensity of carbonyl bands in the 2030–1950 cm−1 region for 773 K reduced samples and for Zn containing samples reduced at lower temperatures suggests that sites responsible for these carbonyl bands may be the active sites in the selective hydrogenation reaction.
Introduction
The selective hydrogenation of organic molecules over heterogeneous catalysts is a reaction that has received significant attention in recent years due to its relevance in producing appropriate reagents for the fine chemicals industry. Of these hydrogenation reactions, those involving α,β-unsaturated aldehydes such as crotonaldehyde has been selected as a test reaction to model systems where more than one reducible functional group is present.1–4 Clearly, the modes of adsorption of the α,β-unsaturated aldehyde and the nature of its preferred adsorption sites are paramount in controlling selectivity in the reaction. Theoretical5,6 and experimental studies7,8 have been conducted using platinum single crystal surfaces in both monometallic and alloyed forms, although fewer studies at a fundamental level have been performed of the adsorption modes on supported metal catalyst surfaces9–11 and fewer specifically deal with catalysts in an SMSI state.11,12 The use of supported catalyst to study adsorption modes involves many complications, among which is that adsorption of the C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C or CO double bond parallel to the surface as a prerequisite to hydrogenation renders the stretching modes IR inactive as a consequence of the surface selection rule. Additionally, the spectra may be dominated by bands due to carboxylates and molecularly adsorbed aldehyde at exposed Lewis acid centers on the surface of the support.10,11 In such cases, the IR spectrum may be dominated by spectator species which make the detection of adsorption on the active catalyst component more difficult. Infrared spectroscopy of adsorbed CO may be used to provide information regarding size and morphology of metallic particles and the extent of interaction with the support, other promoters, and co-adsorbates.13–17 Experiments involving the co-adsorption of CO and 2-butenal (crotonaldehyde) may be used to derive information relevant to the both the metal-support interaction and the interaction of these components with the α,β-unsaturated aldehyde.10,11 Previous studies using co-adsorption have been used to show the presence of electronic effects and redox reactions of crotonaldehyde in cases where the active metal is readily oxidized or reduced and the support may be influential in inducing selectivity of the reaction.10 A recent study12 suggests that the addition of Zn to supported platinum catalysts may increase the metal support interaction and thus modify the catalyst behavior for 2-butenal hydrogenation. In the present study, this same series of catalysts have been studied by FTIR spectroscopy to determine if further evidence is available to support such conclusions.
C or CO double bond parallel to the surface as a prerequisite to hydrogenation renders the stretching modes IR inactive as a consequence of the surface selection rule. Additionally, the spectra may be dominated by bands due to carboxylates and molecularly adsorbed aldehyde at exposed Lewis acid centers on the surface of the support.10,11 In such cases, the IR spectrum may be dominated by spectator species which make the detection of adsorption on the active catalyst component more difficult. Infrared spectroscopy of adsorbed CO may be used to provide information regarding size and morphology of metallic particles and the extent of interaction with the support, other promoters, and co-adsorbates.13–17 Experiments involving the co-adsorption of CO and 2-butenal (crotonaldehyde) may be used to derive information relevant to the both the metal-support interaction and the interaction of these components with the α,β-unsaturated aldehyde.10,11 Previous studies using co-adsorption have been used to show the presence of electronic effects and redox reactions of crotonaldehyde in cases where the active metal is readily oxidized or reduced and the support may be influential in inducing selectivity of the reaction.10 A recent study12 suggests that the addition of Zn to supported platinum catalysts may increase the metal support interaction and thus modify the catalyst behavior for 2-butenal hydrogenation. In the present study, this same series of catalysts have been studied by FTIR spectroscopy to determine if further evidence is available to support such conclusions.
Experimental
Pt/CeO2–SiO2 and PtZn/CeO2–SiO2 catalysts were prepared by impregnation of a 20 wt.% CeO2/SiO2 support using an acetone solution of [Pt(NH3)4](NO3)2 and Zn(NO3)2·6H2O (from Aldrich). A ratio of 10 cm3 of impregnating solution per gram of support was used in both cases. Precursors were selected to avoid the presence of chlorine in the final catalysts. The excess solvent was removed by flowing N2 through the suspension. The remaining solid was dried overnight at 373 K and calcined in air at 673 K for 4 h. A more detailed description of the preparation procedure can be found elsewhere.12 Platinum content was 1.1 wt.% in both samples and the zinc content in the bimetallic catalyst was 0.6 wt.%, as determined by AAS. These values correspond to a Zn/Pt atomic ratio of 1.6. BET surface areas were 211 and 228 m2 g−1 for the Zn-free and Zn containing catalysts, respectively.FTIR experiments were conducted using a quartz infrared cell fitted with CaF2 windows and an external furnace. The IR chamber was glass-blown to a conventional vacuum system with rotary and oil diffusion pumps achieving pressures of down to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ×10−3 N m−2. Catalyst samples were prepared as 16 mm diameter self-supporting disks by pressing 30 mg of loose powder between polished steel dies at 20 MN m−2. Prior to IR measurements, samples were reduced in situ under a hydrogen flow (50 cm3 min−1) at 473, 623 or 773 K for 1 h. After the reduction treatment, samples were out-gassed at the reduction temperature for 20 min to a final pressure of ≈10−5 Torr before cooling to ambient temperature. Pulses of CO and crotonaldehyde were introduced at 298 K using calibrated volumes and an Edwards active strain gauge to measure the size of the pulse. IR spectra were recorded using a PC controlled Perkin-Elmer 1710 FTIR spectrometer operating at 4 cm−1 resolution and averaging 100 scans per spectrum. Crotonaldehyde was purified by repeated distillation under reduced pressure. For the co-adsorption measurements, 25 Torr doses of CO were initially introduced followed by evacuation at 298 K for 20 min prior to exposure to a crotonaldehyde pulse corresponding to low (2.8 μmol gcat−1) and high (41.5 μmol gcat−1) coverage of the surface. These values correspond with 8×10−3
(low) and 0.12 molecules nm−2
(high) for the Zn-free sample and 7.4×10−3 and 0.11 molecules nm−2 for the Zn-containing samples.
×10−3 N m−2. Catalyst samples were prepared as 16 mm diameter self-supporting disks by pressing 30 mg of loose powder between polished steel dies at 20 MN m−2. Prior to IR measurements, samples were reduced in situ under a hydrogen flow (50 cm3 min−1) at 473, 623 or 773 K for 1 h. After the reduction treatment, samples were out-gassed at the reduction temperature for 20 min to a final pressure of ≈10−5 Torr before cooling to ambient temperature. Pulses of CO and crotonaldehyde were introduced at 298 K using calibrated volumes and an Edwards active strain gauge to measure the size of the pulse. IR spectra were recorded using a PC controlled Perkin-Elmer 1710 FTIR spectrometer operating at 4 cm−1 resolution and averaging 100 scans per spectrum. Crotonaldehyde was purified by repeated distillation under reduced pressure. For the co-adsorption measurements, 25 Torr doses of CO were initially introduced followed by evacuation at 298 K for 20 min prior to exposure to a crotonaldehyde pulse corresponding to low (2.8 μmol gcat−1) and high (41.5 μmol gcat−1) coverage of the surface. These values correspond with 8×10−3
(low) and 0.12 molecules nm−2
(high) for the Zn-free sample and 7.4×10−3 and 0.11 molecules nm−2 for the Zn-containing samples.
Prior to determining the catalytic behavior, catalysts were reduced by heating in situ under flowing hydrogen (50 cm3 min−1) to 473, 623 or 773 K at 5 K min−1 and then holding the final temperature for 1 h. The vapor-phase hydrogenation of crotonaldehyde was performed at atmospheric pressure and at a temperature of 353 K. The reduced catalysts (ca. 100 mg) were contacted with a reaction mixture (total flow: 50 cm3 min−1; H2/aldehyde ratio of 26) formed by flowing hydrogen through a thermostabilized saturator (293 K) containing the aldehyde (>99.5% purity, Fluka). The reaction products were analyzed by on line gas chromatography, using a Carbowax 20M 58/90 25–30 m capillary column.
Results
Adsorption of CO on reduced Pt/CeO2–SiO2 and PtZn/CeO2–SiO2 catalysts
FTIR measurements of adsorbed CO on Pt/CeO2–SiO2 catalyst reduced at (A) 473 K, (B) 623 K and (C) 773 K are shown in Fig. 1. In all cases, samples at 298 K were exposed to increasing pressures of CO up to a maximum of 25 Torr followed by evacuation for 40 min. For the sake of brevity, only spectra at 0.5 and 25 Torr are shown. Spectra of the monometallic catalyst reduced at 473 K (A) exhibits a broad band centered at 2067 cm−1 which became more defined on increasing CO pressure. This increased pressure also produces a high frequency shoulder at 2080 cm−1 together with a broad contribution at 2169 cm−1 associated with CO species in the gas phase. A broad maximum of low intensity at ca. 1850 cm−1 due to bridge bound CO was detected at the higher pressure. Evacuation at 298 K red-shifted the 2067 cm−1 band by 5 cm−1 without significant change in intensity. Reduction at 623 K (B) again produced two contributions at 2067 and 2080 cm−1, however the relative intensities were reversed with respect to the 473 K reduced surface with the former band now present as a low frequency shoulder on the latter. The band due to bridge bound CO was very weak under these conditions but grew on increasing the pressure. An increase in the CO pressure also led to increases in intensity of both of the linear features with the lower frequency contribution undergoing the greater increase. This increase in intensity was also accompanied by a blue shift of both bands to 2072 cm−1 and 2086 cm−1. At this maximum pressure a shoulder centered at 2003 cm−1 was also discernible. Evacuation treatment for 40 min produced a red shift of the linear CO bands to 2080 cm−1 and 2065 cm−1. IR spectra (Fig. 1C) of the Pt/CeO2–SiO2 catalyst reduced at the highest temperature (773 K) showed a broad band with a maximum at 2071 cm−1 together with a high frequency shoulder at 2080 cm−1 and some low frequency contribution to the overall band envelope. Those low frequency contributions were more prominent following an increase in CO pressure, this condition also favoring the appearance of the 2003 cm−1 maximum, which was more dominant than for sample reduced at 623 K. Evacuation produced an increase in intensity at 2063 cm−1 accompanied by reduced intensity at 2003 and ca. 1850 cm−1. | ||
Fig. 1 Infrared spectra of a Pt/CeO2–SiO2 catalyst reduced at (A) 473 K, (B) 623 K and (C) 773 K and exposed to (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) 0.5 Torr CO, ( ) 0.5 Torr CO, (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) 25 Torr CO and then (—) outgassed at 298 K for 40 min. ) 25 Torr CO and then (—) outgassed at 298 K for 40 min. | ||
Fig. 2 shows IR spectra for CO adsorbed on the bimetallic PtZn/CeO2–SiO2 catalyst after low (A: 473 K), medium (B: 623 K) and high (C: 773 K) temperature reduction. All spectra exhibit bands in the 2080–2057 cm−1 region which correspond to linear adsorbed CO species on the different metallic sites. After reduction at 473 K and exposure to a low pressure (0.5 Torr) of CO, spectra exhibited a broad band with a maximum centered at 2072 cm−1 together with a shoulder at 2055 cm−1. Exposure to higher CO pressures produced an increase in both contributions to leave a doublet at 2079 and 2065 cm−1 of similar intensities. At this higher pressure, features at ca. 1850 and 2003 cm−1 were also discernible. Note that the latter feature was not present at this reduction temperature for the Zn-free sample. Subsequent evacuation treatment led to a decrease in the intensity of both main peaks and a red shift in both maxima to 2074 and 2061 cm−1. This was accompanied by significant intensity loss at 2000 and 1850 cm−1. A sample reduced at 623 K and exposed to a low CO pressure produced a spectrum with the main feature centered at 2067 cm−1 with associated shoulders at 2046 and 2003 cm−1. An increase in CO pressure shifted the main peak to 2071 cm−1 (with a poorly defined shoulder at 2082 cm−1) and the aforementioned contribution at 2003 cm−1 and the bridge bound band at 1850 cm−1 were both increased in intensity. Subsequent evacuation for 40 min removed adsorbed CO responsible for the high frequency band but the band at 2071 cm−1 was largely unaffected. This treatment also modified the appearance the lower frequency range with a tail of notable intensity between 2050 and 1950 cm−1 with maxima around 2047 and 2027 cm−1. After a high temperature (773 K) reduction treatment the IR spectrum after low CO pressure exhibited two main bands at 2079 and 2065 cm−1 which became more prominent and showed shifts to higher frequencies under higher CO pressures. These two bands were accompanied by several lower frequency contributions in the 2050–1950 cm−1 range which were retained after evacuation at 298 K. Bridge bound CO was less significant for samples reduced at 773 K.
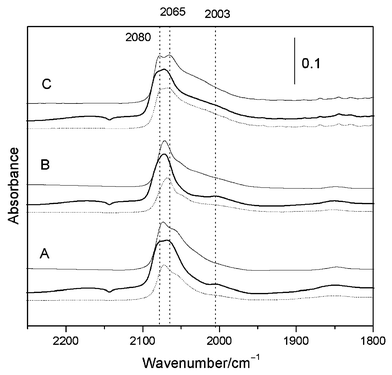 | ||
| Fig. 2 Infrared spectra of a PtZn/CeO2–SiO2 catalyst reduced at (A) 473, (B) 623 and (C) 773 K and exposed to () 0.5 Torr CO, () 25 Torr CO and then (—) outgassed at 298 K for 40 min. | ||
Co-adsorption of CO and crotonaldehyde
Fig. 3 displays IR spectra corresponding to the adsorption of 25 Torr CO on the Zn-free Pt/CeO2–SiO2 catalyst followed by evacuation and the introduction of low and high coverage doses of crotonaldehyde over the CO covered surface. Irrespective of the catalyst reduction treatment (473 K (A), 623 K (B) and 773 K (C)), interaction of crotonaldehyde with the CO covered surface produced a shift to lower wavenumbers of the maxima due to linearly adsorbed CO. Similar observations were made when CO and crotonaldehyde were co-adsorbed on Pt/TiO2 (11). The sample reduced at 473 K (Fig. 3A) and treated with 25 Torr CO followed by evacuation exhibited bands at 2078 and 2062 cm−1 as observed in previous spectra (Fig. 1A). This observation confirms the degree of reproducibility of the experiments. A low coverage crotonaldehyde pulse introduction to the CO covered, 473 K reduced surface, produced a slight shift of both contributions to 2073 and 2059 cm−1, respectively. At higher crotonaldehyde coverage, the intensity of the higher frequency band was most affected while the lower frequency component retained its intensity and its FWHM but was red shifted to 2049 cm−1. A sample reduced at 623 K (B) showed similar behavior. Following CO exposure and brief evacuation, the IR spectrum (Fig. 3B) exhibited two maxima at 2080 and 2067 cm−1 as before (Fig. 1B) together with some low frequency shoulders which gave rise to the observed tail around 2030 cm−1. Exposure to a low coverage pulse of crotonaldehyde produced a decrease in intensity of the high frequency component without significant displacement, whereas the lower frequency band underwent a 5 cm−1 red shift to 2062 cm−1 but with a much lesser intensity loss. This effect was further pronounced for a higher coverage crotonaldehyde pulse where the 2080 cm−1 band intensity was significantly reduced again without appreciable frequency shift while the main maximum was further displaced to 2050 cm−1 without appreciable intensity loss or change in the FWHM. After a high temperature reduction treatment, the Zn-free catalyst exposed to 25 Torr CO exhibited an intense band at 2071 cm−1 together with a shoulder at 2083 cm−1. Exposure to a low coverage crotonaldehyde pulse produced minor changes to the spectra with a slight decrease in the intensity of both bands and a red-shift in the lower frequency component by 2 cm−1. However, exposure to higher crotonaldehyde vapor pressure produced more significant changes in the spectrum with the high frequency component being significantly attenuated without undergoing a shift in frequency and the lower frequency component was now present at 2058 cm−1 and exhibited marginally less intensity. | ||
| Fig. 3 Infrared spectra of a Pt/CeO2–SiO2 catalyst reduced at (A) 473, (B) 623 and (C) 773 K, exposed to 25 Torr CO followed by outgassing at 298 K for 20 min () and exposure to a low coverage pulse of crotonaldehyde (—) and a high coverage crotonaldehyde pulse (). | ||
Fig. 4 shows spectra obtained following the co-adsorption of CO and crotonaldehyde over the PtZn/CeO2–SiO2 catalyst after reduction treatment at 473 K (A), 623 K (B) and 773 K (C). As observed in the case of the Zn-free catalyst, and irrespective of the reduction treatment applied, interaction of crotonaldehyde with a CO covered surface led to a red shift of the overall band envelope of linearly adsorbed CO. Additionally, although less readily observed due to the relatively low intensity, addition of crotonaldehyde led to a red shift of the band centered at ca. 1850 cm−1 due to bridge bound carbonyls. The sample reduced at 473 K and pre-covered with CO exhibited a broad band centered at 2068 cm−1 together with considerable asymmetry towards low frequencies. Exposure to a low coverage pulse of crotonaldehyde produced a definition of this broad band into two contributions at 2071 and 2057 cm−1, with the former suffering the greater loss in intensity. At a higher vapor pressure of crotonaldehyde there was a further intensity decrease in the high frequency component without significant shift in frequency while the lower frequency maximum was now located at 2042 cm−1 and exhibited a FWHM (79 cm−1) which was of greater magnitude than prior to crotonaldehyde exposure (FWHM=50 cm−1). The tail on this broad band presented a poorly resolved maximum at 2008 cm−1. Similar experiments performed on a sample reduced at 623 K showed equivalent behavior. The sample exposed to CO alone exhibited a maximum intensity at 2067 cm−1 together with a shoulder at 2052 cm−1. Both contributions were better defined after the introduction of a low coverage pulse of crotonaldehyde which led to a slight red shift of the overall band envelope due to linearly adsorbed CO. Exposure to a high coverage pulse of crotonaldehyde produced a significant decrease in intensity of the high frequency component (ca. 2067 cm−1) band together with a shift to lower wavenumber of the main peak. As observed for the 473 K reduced sample (Fig. 4A), this broad maximum was centered at 2042 cm−1 and showed evidence for a low frequency contribution around 2008 cm−1 which contributed to the low frequency shoulder tail. A reduction treatment at the highest temperature (773 K) on the Zn containing catalyst followed by exposure to CO produced a maximum with evidence for two components centered at 2069 and 2078 cm−1, and a long tail between 2050 and 1950 cm−1. Exposure of the CO covered surface to low dose of crotonaldehyde produced only minor changes in the overall band envelope but with an indication of reduced intensity of the main peaks and gained intensity in the tail. This modification was enhanced by the addition of further crotonaldehyde which left the main maximum at 2072 cm−1 with an equivalent intensity as the component which made up the tail, now centered at ca. 2030 cm−1.
 | ||
| Fig. 4 Infrared spectra of a PtZn/CeO2–SiO2 catalyst reduced at (A) 473, (B) 623 and (C) 773 K, exposed to 25 Torr CO followed by outgassing at 298 K for 20 min () and exposure to a low coverage pulse of crotonaldehyde (—) and a high coverage crotonaldehyde pulse (). | ||
Adsorption of crotonaldehyde
Figs. 5 and 6 display spectra of crotonaldehyde exposed at increasing vapor pressures to a representative Pt/CeO2–SiO2 and Pt/Zn/CeO2–SiO2 catalysts, respectively, both reduced at 773 K. The third and last spectra in each case represent the “low” and “high” coverage pulses, respectively. All catalysts displayed very similar spectra with the exception of the 473 K reduced Zn-free sample which showed no band at 1560 cm−1. This feature was common to spectra recorded of the Zn containing samples regardless of reduction temperature and the Zn-free sample when reduced at 623 or 773 K. Other features were bands at 1678 (sh), 1668, 1643, 1446, 1399, 1378 and 1307 cm−1. These features grew with increased exposure at low vapor pressures indicating that these were all modes of a single adsorption mode which dominated at low coverage. The only exception was the low temperature (473 K) reduced Zn-free sample where the dominant feature at low coverage was located at ca. 1630 cm−1 but which became dominated by the species giving the bands listed above features as coverage was increased. Bands at 1446 and 1378 cm−1 are due to the asymmetric and symmetric deformations of the methyl group. The 1307 cm−1 band corresponds with the –CH in-plane deformation (1303 cm−1 in the liquid phase spectrum). The 1643 cm−1 band is due to the CC stretching mode, while the band at 1668 cm−1 is due to the νCO. The red shift in the latter and the unperturbed nature of the former indicate that the dominant mode of adsorption at low coverage involved the molecule adsorbed via the carbonyl rather than the vinyl function, probably ligated to exposed cationic sites acting as Lewis acid centers in the support surface. However, as the XPS atomic ratios12 were only ca. 8.3 ∶ 1 for Ce ∶ Pt, and neglecting the possibility of chemisorbed aldehyde on the exposed silica surface, it is quite possible that crotonaldehyde adsorbed at exposed Pt sites contributed to the IR spectra (Fig. 5). The growth of the feature at 1678 (sh) cm−1 at higher coverage can be attributed to the νCO of the aldehyde, again adsorbed via the carbonyl function, but at weaker adsorption sites, possibly involving hydrogen bonding interactions with surface hydroxyl species. An additional feature, also observed at higher coverage was a band at 1602 cm−1 which was very clearly observed for the Zn containing catalyst reduced at 773 K (Fig. 5), less clear, but still a resolved feature for Zn-free catalyst reduced at 773 K (Fig. 6), and absent for both samples reduced at 473 and 623 K. This feature may be attributed to νCC of a π bound species.
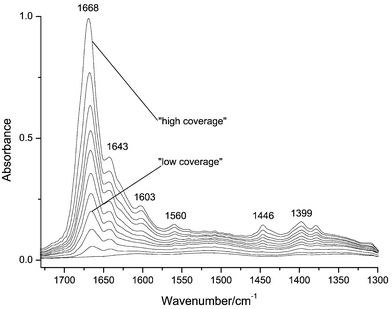 | ||
| Fig. 5 FTIR spectra of PtZn/CeO2–SiO2 catalyst reduced at 773 K and exposed to increasing vapor pressures of crotonaldehyde at 298 K. | ||
 | ||
| Fig. 6 FTIR spectra of Pt/CeO2–SiO2 catalyst reduced at 773 K and exposed to increasing vapor pressures of crotonaldehyde at 298 K. | ||
Hydrogenation of crotonaldehyde
Table 1 reports the activity and selectivity for crotonaldehyde hydrogenation at 353 K over Pt/CeO2–SiO2 and PtZn/CeO2–SiO2 reduced in flowing H2 at different temperatures (473, 623 and 773 K) for 1 h. The initial data are taken after 4 min on stream, when the carbon balance was achieved. Prior to this, a given amount of reactant and products were being adsorbed on the catalysts and then, no reliable measurements could be done. The comparation between the initial data (t=4) and those measured after 200 min on stream the degree of catalyst deactivation and its effect on selectivity to be evaluated.
| Selectivity (%) | ||||||
|---|---|---|---|---|---|---|
| Catalyst | Reduction temperature/K | Time on stream/min | Activity/μmol (g Pt)−1 s−1 | Butanal | Crotyl alcohol | Conversion (%) |
| Pt/CeO2–SiO2 | 473 | 4 | 204 | 92 | 2 | 10 |
| 200 | 157 | 95 | 0 | 8 | ||
| 623 | 4 | 227 | 72 | 9 | 11 | |
| 200 | 144 | 94 | 2 | 7 | ||
| 773 | 4 | 189 | 56 | 40 | 8 | |
| 200 | 83 | 88 | 9 | 4 | ||
| PtZn/CeO2–SiO2 | 473 | 4 | 55 | 74 | 22 | 2 |
| 200 | 20 | 85 | 5 | 1 | ||
| 623 | 4 | 178 | 58 | 35 | 8 | |
| 200 | 25 | 84 | 10 | 1 | ||
| 773 | 4 | 667 | 63 | 15 | 27 | |
| 200 | 47 | 78 | 22 | 2 | ||
The Zn-free samples did not show a great deal of sensitivity to the reduction temperature. Activity increased slightly when passing from 473 to 623 K and then decreased again slightly after reduction at 773 K. However, the selectivity to crotyl alcohol was strongly affected, mainly after reduction at this highest temperature. It should be noted that selectivity to butanol was nil, and to light hydrocarbons was lower than 5% in all cases. The increase in selectivity to crotyl alcohol with increasing reduction temperature indicates the creation of active sites on which the hydrogenation of the carbonyl bond is carried out at the expense of sites which are active for the hydrogenation of the CC bond. Similar results were obtained for Pt/TiO2 when reduced at temperatures between 473 and 773 K11 although, in that case, the catalyst was strongly deactivated after reduction at 773 K. The Zn-containing catalyst, on the other hand, showed both increased activity and crotyl alcohol selectivity as the reduction temperature was increased. The selectivity to crotyl alcohol was significant here even after reduction at only 473 K. The lower selectivity for the PtZn catalyst reduced at 773 K is related to the high amounts of butanol produced during the initial stages of reaction. Indeed, at t=10 min the selectivity to butanol was only 3% and the selectivity to crotyl alcohol had increased to 36%. All catalysts showed a rapid deactivation with time on stream but reached a stable conversion level after ca. 75 min. Results at t=200 are also shown in Table 1, where it can be seen that the degree of deactivation increased with reduction temperature, this effect being more pronounced for the Zn-containing catalyst.
Discussion
Adsorption of CO
Spectra of adsorbed CO in the region characteristic15 of linearly adsorbed species show three main characteristics independent of the presence of Zn and the reduction temperature. These are: a high frequency component at 2080 cm−1, often present as a shoulder or an unresolved feature, a band at ca. 2065 cm−1 often appearing as the most dominant feature, and a tail between 2050 and 1950 cm−1, probably containing many distinct contributions but mostly showing a maximum at 2003 cm−1. Each will be discussed in turn, firstly in terms of what they reveal regarding the characteristics of the two catalysts and then, in the second section in terms of what they reveal regarding the interactions of the active catalyst components with the crotonaldehyde. The first band corresponds with a frequency at which the dominant maxima were found for Pt/TiO2 catalysts11 although in that study a further, higher frequency contribution at 2094 cm−1 was observed which was absent in this study. The 2094 cm−1 band was assigned to CO adsorbed on close-packed terrace sites such as {111} patches where the carbonyls were exposed to extensive coupling interactions. The band at 2084 cm−1 was attributed11 to carbonyls on less extended terrace sites and/or where the metal atoms were in less densely packed arrangements such as in {100} type facets and such an assignment is accepted here to explain the highest frequency component observed for both Pt/CeO2–SiO2 and PtZn/CeO2–SiO2. The absence here of a feature at ca. 2094 cm−1 could be interpreted in terms of the dominance of more spherical Pt particles over CeO2–SiO2 while flat-raft like particles with extended flat surfaces were favored over the titania support. Copper was also envisaged to prefer a two-dimensional raft-like distribution when supported on titania.10 In addition to the progressive loss of CO and hydrogen chemisorption as a function of increasing reduction temperature, evidence for the formation of an SMSI state for Pt/TiO2 catalysts was the progressive loss of the sites giving the 2094 cm−1 band.11 Spectra for the Zn-free Pt/CeO2–SiO2 sample show an increase in the contribution made by the 2080 cm−1 feature as the reduction temperature was increased from 473 to 673 K (Fig. 1A and B); however, the band was very weak for the 773 K reduced sample despite the overall increase in area of the overall band envelope. Zn containing Pt/CeO2–SiO2 on the other hand showed no overall increase in band envelope on increasing the reduction temperature from 623 to 773 K, however, in contrast to Zn-free sample, the band at 2080 cm−1 was still present as an intense feature for the 773 K reduced sample. One interpretation, made on the basis of previous results for Pt/TiO211 would be that the loss or depletion of the 2080 cm−1 feature in Pt/CeO2–SiO2 at the highest reduction temperature was the consequence of coverage of the sites responsible by sub-oxides of cerium although higher temperatures are usually required for the formation of an SMSI state for ceria than titania.18 In applying the same reasoning to the Zn containing sample, it would have to be argued that the presence of Zn, present as the oxide, hindered the platinum from entering into such a state given the strong presence of the 2080 cm−1 band for this sample even after 773 K reduction. An alternative suggestion would be that the presence of Zn hindered a morphological transition of the Pt catalysts between 623 and 773 K which in its absence resulted in a change in the distribution of the exposed sites such that the centers responsible for the 2080 cm−1 band became less significant and were replaced by sites giving rise to CO absorption bands at a lower frequency. Arguments against a significant change in particle size/shape and inherent changes to morphology for the Pt/CeO2–SiO2 catalysts as the reduction temperature was increased was the invariance in the Pt/Si atomic ratios obtained by XPS for 473 and 773 K reduced samples.12 The balance of the argument would be that the presence of Zn hinders processes which lead to the loss of the sites giving the 2080 cm−1 band and which occur in the absence of Zn when the Pt/CeO2–SiO2 catalysts are reduced between 623 and 773 K.One further point to add regarding the sites giving the 2080 cm−1 band involves the proposal12 that 773 K reduction of PtZn/CeO2–SiO2 catalysts leads to the formation of a Pt–Zn “alloy.” FTIR results here are not consistent with such a proposal. The incorporation of regular arrays of Zn atoms into an otherwise contiguous array of Pt atoms would be expected to have a significant effect on the frequency of νCO. Such carbonyl species when forming part of an extended array of molecules with similar singleton frequency as its nearest neighbors exhibit high stretching frequencies mainly as a consequence of the number of dipole–dipole coupling interactions. Dilution of such an array by the addition of Zn atoms, assuming the absence of an exactly compensating electronic effect by Zn, would lead to reduced frequency of the Pt carbonyls exhibiting the 2080 cm−1 band, a result which is not observed for the 773 K reduced PtZn/CeO2–SiO2 catalyst.
Assuming that the frequency of the Pt carbonyls are mainly a consequence of the coordination number on the adsorbing atom and the number of dipole–dipole interactions with neighboring adsorbed carbon monoxide molecules rather than being dominated by the influence of support-metal interactions, then the band appearing at ca. 2065 cm−1 can be attributed as before11 to sites of lower coordination such as atoms located at edges and kinks and sites at the peripheries of extended facets.15,16,19 Support for the initial statement is made above where it is reasoned that the particles here may be spherical (or hemispherical) rather than raft-like, the latter favoring metal–support interaction. Further evidence that the particles are spherical/hemispherical in nature come from the fact the 2065 cm−1 band is the dominant feature in the majority of the spectra recorded here but higher frequency carbonyl bands dominated spectra of similarly loaded Pt/TiO2 catalysts where flatter particles with more extended facets were thought to dominate. Although the 2065 cm−1 feature already dominated spectra of CO adsorbed on the samples employed here, intensity transfer from low frequency to high frequency species11,14 as a consequence of dipole coupling interactions between carbonyls at adjacent but dissimilar sites, is likely to result in the a concomitant loss of intensity at 2065 cm−1 and equivalent intensity gain at 2080 cm−1 assuming that the two types of sites are both present within the same particle. The observed intensity at 2065 cm−1 might therefore underestimate (in relative terms) the number of sites present although in a similar manner, this band intensity might be enhanced by transfer from yet lower frequency maxima. The formation of inter-metallic compounds involving Pt and an additional metal with a lower enthalpy of sublimation is expected to produce particles in which the other metal, Zn in this case, should occupy the higher energy (i.e. lower coordinated sites) to reduce the overall surface free energy of the particle. Should reduction treatment at 773 K favor12 the formation of such a bimetallic cluster, then the expectation in terms of modified IR spectrum of adsorbed CO would be to diminish the contribution of the 2065 cm−1 feature. Comparison of spectra in Fig. 2A and 2C does not show such a tendency and thus provides further evidence against the formation of bimetallic clusters under these conditions.
Unlike spectra of CO adsorbed on single crystal or extended surfaces, spectra of linearly adsorbed CO on supported Pt always exhibit a main band which displays considerable asymmetry towards lower frequencies11,14–16,19 indicating the presence of sites which are a unique feature of a supported catalyst. This was also a feature for the Pt/CeO2–SiO2 catalyst (with or without Zn) studied here. Although the range of frequencies was similar for Zn-containing and Zn-free catalysts (to 1950 cm−1), the intensity was of significance for both the 773 K, and in particular, for the Zn-containing sample (Fig. 2C). An additional component of this tail was a resolved feature at 2003 cm−1. This was not detected in previous studies of Pt/CeO219 although the maximum reduction temperature employed in that study was 673 K. Spectra here for the Zn-free system are not inconsistent with this result as the 2003 cm−1 feature was only observed as a resolved feature for sample reduced at 773 K. On the other hand, this feature was common to spectra of the Zn containing catalysts for all three reduction temperatures. One plausible interpretation is that the band is due to the Pt bound CO where the oxygen interacts with exposed, possible reduced exposed cerium ions, in the support surface. Such sites would only be created at the metal particle-support interface and would be most abundant in cases where small particles were present or where isolated Pt atoms were distributed over the support. The appearance of the 2003 cm−1 maximum irrespective of reduction temperatures for catalyst containing Zn, but only after 773 K reduction for Zn-free catalysts and an assignment involving reduced, exposed Ce sites is supported by TPR results.12 These experiments show12 that Pt is reduced below 450 K, which is consistent with the failure to detect bands here between 2150 and 2100 cm−1 which are characteristic of CO adsorbed at oxidized platinum sites.15,16,19,20 A second reduction peak, attributed to surface reduction of ceria and centered at 640 K was only complete for Zn-free samples at 700 K so only the IR experiments performed on a 773 K reduced sample would involve an extensively reduced ceria surface. This peak was absent for the Zn containing sample12 but was replaced by a broadened first peak where both the Pt and the surface ceria was reduced in a step with a maximum at 377 and shoulder at 447 K. i.e. in the case of the Zn containing catalyst, IR experiments performed at all reduction temperatures involved predominantly reduced ceria surfaces. In spite of this surface reduction of ceria, samples, even after 773 K reduction, did not show clear signs of an SMSI state, which, in the case of Pt/TiO2 catalysts11 was evidenced by significantly reduced CO uptake and a correspondingly smaller IR band envelope. Results are consistent with recent findings for model Pt/CeO2 systems18 where temperatures above 800 K were required before modifications to the CO chemisorption properties were observed.
A proposed scenario consistent with enhanced ceria reducibility in the presence of Zn12 and the possible role of ZnO in suppressing the onset of an SMSI state (loss of 2080 cm−1 band) would involve a bimodal distribution of particles. Larger particles (giving the 2080 and much of the 2065 cm−1 band) are located on deposits of ZnO where they are effectively isolated from the ceria component. Similar particles are present for Zn-free sample but these are in direct interaction with the ceria support and show some signs (low activity in reaction, loss of the 2080 cm−1 band) of entering into the initial stages of an SMSI state after 773 K reduction. Ceria reduction at lower temperatures, facilitated by the presence of reduced Zn rather than reduced Pt for the Pt–Zn catalyst does not lower the onset temperature for the SMSI state of the larger particles as they are isolated from the reduced ceria. Smaller clusters of Pt and even isolated Pt atoms, are in direct contact with areas of ceria, however the creation of Pt–Ce3+ interface sites where tilted CO molecules are adsorbed (2003 cm−1 band) is only possible when reduction of the ceria surface occurs, a process which is apparently12 facilitated by the presence of zinc.
Note that although the close proximity of Pt and Zn is envisaged in the above model, the suggestion12 that Pt-Zn alloy formation occurs following 773 K reduction is not consistent with the IR results obtained. Although Pt-Zn alloy formation has been observed for Pt/ZnO catalysts reduced at temperatures as low as 473 K21–23 one would expect considerable differences between spectra of the 773 K reduced samples in the presence and absence of Zn. These differences, such as reduced overall band intensity,23 lower frequency of the νCO maximum, invariance in νCO as a function of CO coverage and loss of intensity of bands due to bridge-bonded species as a result of isolation of Pt sites,22 were not apparent.
Adsorption of crotonaldehyde
With the exception of the 473 K reduced Zn-free sample, all other catalysts exhibited a weak feature at 1560 cm−1 which can be attributed to νas(COO) vibrations of adsorbed crotonate anions. Carboxylate species were not formed at low surface coverage and appeared to be only a minority species or absent for samples with limited or no surface ceria reduction (e.g. Zn-free 473 K reduced sample). As in the case of Cu/TiO2,10 the formation of carboxylate species was facilitated by an increase in sample temperature. The dominant mode of adsorption involved interaction of the carbonyl group acting as a Lewis base with exposed surface sites. These were probably mainly exposed Ce4+ sites as the band appeared for all samples regardless of reduction temperature and for samples with no evidence12 of significant surface reduction. In all cases the effect of increasing coverage shifted the maximum from 1664–1668 cm−1. At highest coverages, the carbonyl group was involved in weaker interactions, probably involving hydrogen bonds with exposed hydroxyls. As indicated above, as the Ce ∶ Pt XPS atomic ratios were only ca. 8.3 ∶ 1 it is possible that the spectra contained contribution from crotonaldehyde adsorbed at exposed Pt sites. However if crotonaldehyde favored a di-σco adsorption arrangement as in the case of Pt {111}3 neither the νCO nor the νCC would be observed by IR, unless these were adsorbed at interfacial sites where the surface selection rule would no longer apply. In any case, adsorption on the support clearly made the major contribution, given the limited displacement of CO which occurred from the metal under conditions which gave intense spectra due to adsorbed crotonaldehyde shown in the last spectrum in the series shown in Figs. 5 and 6 and representing the “high coverage” pulse. The 473 K reduced Zn-free catalyst gave evidence that the σ-complex involving ligation via the carbonyl O-atom was not the major species at low coverage. The absence, or low intensity of the carbonyl stretching mode under these conditions and the perturbation of the νCC would be consistent with the π di-σco species proposed by Englisch et al.3 for adsorption at interface sites. However, as adsorption under these conditions (low coverage pulse) displaced very little CO from the surface (Fig. 4A) it is unlikely in this case to involve adsorption sites located on the metal surface. The difference between the dominant low coverage mode of adsorption over 473 K reduced Zn-free samples and the others, is probably a consequence of the low extent of surface reduction12 experienced by this sample.
Co-adsorption
Previous studies10,11 involving co-adsorption of CO and crotonaldehyde over titania supported metal catalysts have shown that several effects are observed which depend on the nature of the metal adsorbent. For Cu/TiO2 catalysts, the principle effects of crotonaldehyde on adsorbed CO were summarized10 as; a geometric blocking effect which reduced the surface coverage of CO, an electronic effect due to the electron donating nature of crotonaldehyde acting as a Lewis base and a redox effect which resulted in the oxidation of crotonaldehyde and the reduction of Cu(I) to Cu(0). On the other hand, Pt/TiO2 catalysts exhibited effects which were interpreted11 in terms of the displacement of CO from the weakest adsorption sites which as a consequence of reduced dipole coupling interactions between CO on different adsorption sites, diminished the intensity transfer phenomenon14 which then led to enhanced intensity at lower frequencies. In both cases the titania support was considered to play a significant role in the overall surface chemistry although only in the case of supported platinum catalyst11 was it envisaged as acting in a manner characteristic of an SMSI effect. Supported copper did not display the usual characteristics of a catalyst in an SMSI state.10Unlike Cu/TiO2,10 the possibility of redox behavior during co-adsorption involving crotonaldehyde can be ruled out. Although XPS results12 provide some evidence for Pt states which are reduced to a greater extent following high temperature treatment, the IR results provide no evidence for the generation of new, additional sites following adsorption of crotonaldehyde. The complete band envelope for adsorbed CO in the presence of the aldehyde fell within the same overall wavenumber range as in the presence of CO alone (although the distribution of intensity at each wavenumber was changed). Neither do results indicate that the presence of aldehyde acts only to reduce the surface coverage of COad (geometric effect) as this would eventually lead to spectra akin to those observed at low coverage with a band of reduced intensity right across the range of νCO, i.e. the observed effect would be akin to a desorption experiment in the presence of CO alone.
If a Pt dispersion of 50% is assumed, then sufficient aldehyde molecules would be present in the initial pulse to interact with 10% of all exposed Pt atoms assuming a 1 ∶ 1 configuration. Addition of the first pulse of crotonaldehyde had very little effect on the spectra, resulting in slight loss at the maxima, and an equivalent gain in intensity on the low-frequency tail. It is clear that the vast majority of the molecules in the initial dose are adsorbed by the support, most likely at exposed Lewis acid centers of the support as suggested by spectra in Fig. 5. Increased amounts of aldehyde (equivalent to 1.5 molecules of aldehyde per Pt surface atom at 50% dispersion) caused a red shift in the band envelope of both linear and bridged species but without significant loss in the overall integrated intensity. As no obvious loss in bridge-bonded species accompanied this process, it is unlikely that the loss of CO adsorption sites by displacement by crotonaldehyde is compensated by conversion of bridging to linear sites. It is possible however that loss of CO adsorption sites without significant loss of band intensity is a consequence of compensation by the creation of modified sites (e.g. with aldehyde at an adjacent site) where the adsorbed CO has a higher absorption coefficient. Again this scenario is unlikely to be the dominant effect as exact compensation would appear somewhat fortuitous.
Co-adsorption of CO with unsaturated hydrocarbons or Lewis bases on supported platinum catalysts has previously been shown to produce red-shifts in the maxima due to νCO24,25 which has been interpreted in terms of electronic effects which are expected to increase the extent of back donation into the carbon monoxide π* orbitals, thus reducing the carbon-oxygen bond order. These shifts were ca. 40 cm−1 for benzene,24,25 37 cm−1 for ethene25 and 67 cm−1 for ammonia.25 It is tempting, therefore to interpret shifts in the main linear carbonyl bands (listed for increasing reduction temperature) of 14, 18 and 15 cm−1 for Zn-free catalyst and 21, 22 and ca. 30 cm−1 for Zn containing catalyst to electronic effects induced by the presence of crotonaldehyde. However, Stoop et al.26 using 12CO and 13CO mixtures, were able to show that co-adsorbed ethene exhibited only a very small electronic effect and that the majority of the ca. 35 cm−1 shift in νCO was the consequence of separation of the dipoles (i.e. a geometric effect). One scenario which would account for the limited electronic effects26 and the red-shift in νCO in the presence of co-adsorbates24,25 would be that aldehyde (or unsaturated hydrocarbon) was adsorbed at the interface sites, primarily on the support sites but bridging the interface such that CO at the boundary Pt atoms were displaced. The groups of Vannice1,27 and Lercher3 both envisage these locations as being the active sites for hydrogenation based on activities using different supports and varying reduction temperatures. While not at odds with the model of the active site, FTIR results of the co-adsorption studies here are not consistent with this picture of initial CO displacement. Should the aldehyde adsorb at exposed cation sites in the support–metal interface, then the presence of the adsorbed aldehyde would eliminate any interaction between CO molecules which were simultaneously interacting with Pt atoms and exposed cationic sites at the interface. As argued above, such sites might be responsible for the resolved feature at 2003 cm−1. This band, however, was absent following evacuation at 298 K and so the adsorption of aldehyde at such sites is not proven experimentally. However, what is shown quite clearly both here and in previous studies of supported Pt catalysts11 is that these carbonyls giving rise to the highest frequency component of the band envelope are those which are initially displaced by the aldehyde. These species exhibiting high νCO are unlikely to be located at interface sites as in such a location they would experience fewer dipole interactions and would exhibit lower stretching frequencies than their central neighbors located on metal atoms within an equivalent facet, but surrounded by more nearest neighbor dipoles. Note that an electronic effect induced by aldehyde adsorbed on the support and influencing the metal support interaction is also unlikely to explain results since a shift in the entire band envelope without loss of intensity at a specific frequency would be expected, or, if only the smaller particle are influenced, then only the lower frequency carbonyls would be shifted. This is not consistent with the observed results where a shift in the whole band envelope was observed with selective loss in intensity at the highest frequency.
The loss of the highest frequency carbonyls as aldehyde is introduced reflects the relative ease by which these CO molecules are displaced and is consistent with the fact that these sites are the last to be filled on increasing coverage (Figs. 1 and 2). As these sites (see above) are due to CO on more extended facets, displacement of CO results in a shift to lower frequency as the number of dipole–dipole interactions is reduced. This does not, however, infer anything about the most active site for crotonaldehyde adsorption/reaction. The selective displacement of CO from these sites rather than a more random desorption process which accompanies outgassing, maintains regionally high densities of CO in other facets of the metal crystallite. The elimination from adjacent facets of carbonyls which exhibit higher stretching frequencies, eliminates intensity transfer14 resulting in the growth in the lower frequency region of the spectra shown in Figs. 3 and 4. Note that the separation (Δν∼30 cm−1) between the frequency at which intensity is lost (ca. 2080 cm−1 in Fig. 3 or 2070 cm−1 in Fig. 4) and where the maximum is gained (ca. 2050 cm−1 in Fig. 3 or 2042 cm−1 in Fig. 4) is equivalent to the expected separation between a molecule on a terrace and a defect site14,28 and is small enough to allow dipole coupling interactions.14 The large FWHM of the Zn containing catalysts in the presence of crotonaldehyde compared to Zn-free sample, is a consequence of the greater intensity contribution at low frequencies with the initial CO band envelope although both samples show a return to background absorption at the same frequency (ca. 1950 cm−1).
One possible link between the spectroscopic characterization and reaction data, would be that high temperature treatments or the addition of Zn result in the formation of small clusters of Pt atoms with low coordination and as a consequence of their size, have a large interface zone. These sites are evidenced by the low frequency tail exhibited by all samples here but of greater significance for the 773 K reduced samples and specifically, the Zn-containing samples. This spectroscopic feature is not present for single crystal surfaces and so can be assigned to sites which are a unique feature of a supported catalyst. The small size of the cluster and their inherent close contact with support makes these sites highly sensitive to pretreatment effects and the addition of modifiers to the carrier. Furthermore, as the presence of Zn would appear to enhance the reducibility of ceria,12 this increased number of Ce3+ sites, when located in the interfacial zone close to platinum atoms could be expected to facilitate activation of the aldehyde carbonyl bond and lead to the observed enhanced yields of crotyl alcohol. Although qualitatively, agreement exists between the activity and the magnitude of the band intensity between 2050 and 1950 cm−1, an attempt to show a quantitative relationship between integrated intensity of the νCO band in this region and the yield to crotyl alcohol proved unsuccessful, probably as a consequence of the number of other contributing factors, such as activity of other centers (selective or otherwise) and deactivation of sites as a function of time on stream.
Conclusions
IR spectra of high temperature treated Zn containing and Zn-free PtCeO2/SiO2 samples do not, unlike Pt/TiO2 catalysts, show significant signs of an SMSI state although enhanced selectivity to the unsaturated alcohol was observed for such samples and for low temperature reduced Zn containing samples. The presence of Zn facilitated the reduction of surface ceria, thereby augmenting the potential number of Ce3+-Pt metal interface sites, particularly in cases where small Pt particles were located in ceria rich zones of the support. Such Pt sites were defined by low frequency (2030–1950 cm−1) carbonyl species. The presence of Zn enhanced the number of sites that gave such low frequency carbonyl species, lending to the proposal that such Pt atoms may be the active sites for the hydrogenation reaction. Co-adsorption experiments provide limited evidence for the active sites as spectra are dominated by dipole coupling effects. Introduction of crotonaldehyde to a CO covered surface leads to CO displacement from the weakest adsorption sites which had the effect of enhancing intensity at lower frequencies as a consequence of the removal of intensity transfer phenomena.Acknowledgements
We thank the M.E.C. (Spain) for a travel grant (to J.S.A) which financed a trip to the University of Dundee during which the FTIR measurements were conducted. Financial support from C.I.C.Y.T (Project BQU 2000-0467) is also gratefully acknowledged.References
- M. A. Vannice and B. J. Sen, J. Catal., 1989, 115, 65 CrossRef CAS.
- H. Yoshitake and Y. Iwasawa, J. Chem. Soc., Faraday Trans., 1992, 88, 503 RSC.
- M. Englisch, A. Jentys and J. A. Lercher, J. Catal., 1997, 166, 25 CrossRef CAS.
- B. Coq, P. S. Kumbhar, C. Moreau and M. G. Warawdekar, J. Mol. Catal., 1993, 85, 215 CrossRef CAS.
- F. Delbecq and P. Sauset, J. Catal., 1995, 152, 217 CrossRef.
- F. Delbecq and P. Sauset, J. Catal., 1996, 164, 152 CrossRef CAS.
- F. Beccat, J. C. Bertolini, Y. Gautier, J. Massadier and P. Ruiz, J. Catal., 1990, 126, 451 CrossRef CAS.
- T. Birchem, C. M. Pradier, Y. Berthier and G. Cordier, J. Catal., 1994, 146, 503 CrossRef CAS.
- J. E. Bailie, J. A. Anderson, C. H. Rochester, N. N. Richardson, N. Hodge, J. G. Zhang, A. Burrows, C.J. Kiely and G.J. Hutchings, Phys. Chem. Chem. Phys., 2001, 3, 4113 RSC.
- F. Coloma, B. Bachiller-Baeza, C. H. Rochester and J. A. Anderson, Phys. Chem. Chem. Phys., 2001, 3, 4817 RSC.
- F. Coloma, J. M. Coronado, C. H. Rochester and J. A. Anderson, Catal. Lett., 1998, 51, 155 Search PubMed.
- J. Silvestre-Albero, F. Rodríguez-Reinoso and A. Sepúlveda-Escribano, J. Catal., 2002, 210, 127 Search PubMed.
- R. A. Dalla Betta, J. Phys. Chem., 1975, 79, 2519 CrossRef CAS.
- P. Hollins, Surf. Sci. Rep., 1992, 16, 51 CrossRef.
- C. De la Cruz and N. Sheppard, Spectrochim. Acta, Part A, 1994, 50, 271 CrossRef.
- J. A. Anderson, Catal. Lett., 1992, 13, 363 Search PubMed.
- C. Hippe, R. Lamber, G. Schulz-Ekloff and U. Schubert, Catal. Lett., 1997, 43, 195 Search PubMed.
- D. R. Mullins and K. Z. Zhang, Surf. Sci., 2002, 513, 163 CrossRef CAS.
- T. Jin, Y. Zhou, G. J. Mains and J. M. White, J. Phys. Chem., 1987, 91, 5931 CrossRef CAS.
- J. A. Anderson, J. Chem. Soc., Faraday Trans., 1992, 88, 1197 RSC.
- M. Consonni, D. Jokie, D. Y. Murzin and R. Touroude, J. Catal., 1999, 188, 165 CrossRef CAS.
- F. Boccuzzi, A. Chiorino and G. Ghiotti, Surf. Sci., 1989, 209, 77 CrossRef CAS.
- F. Boccuzzi, A. Chiorino, G. Ghiotti, F. Pinna, G. Strukul and R. Tessari, J. Catal., 1990, 126, 381 CrossRef CAS.
- J. M. Basset, G. Dalmai-Imelik, M. Primet and R. Mutin, J. Catal., 1975, 37, 22 CrossRef CAS.
- M. Primet, J. Catal., 1984, 88, 273 CrossRef CAS.
- F. Stoop, F. J. C. M. Toolenaar and V. Ponec, J. Catal., 1982, 73, 50 CrossRef CAS.
- M. A. Vannice, Top. Catal., 1997, 4, 241 Search PubMed.
- R. G. Greenler and R. K. Brandt, Colloids Surf., 1995, 105, 19 Search PubMed.
| This journal is © the Owner Societies 2003 |