Electronic spectroscopy of the CaCCCH3 and SrCCCH3 free radicals
Gary K.
Corlett
,
Ketan
Patel
,
Martyn D.
Wheeler
and
Andrew M.
Ellis
*
Department of Chemistry, University of Leicester, University Road, Leicester, UK LE1 7RH
First published on 18th November 2002
Abstract
The first spectroscopic observation of the free radicals CaCCCH3 and SrCCCH3 is reported. Vibrationally-resolved laser-induced fluorescence spectra, which were recorded under supersonic jet conditions, reveal two band systems for each molecule which have been assigned to the Ã
2E–![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 2A1 and
2A1 and ![[B with combining tilde]](https://www.rsc.org/images/entities/char_0042_0303.gif) 2A1–
2A1 electronic transitions. The vibrational structure in each system is limited to a short progression in the metal–carbon stretching mode. The Ö origin is blue-shifted relative to the monoacetylides of Ca and Sr. Furthermore, in contrast to CaCCH and SrCCH, where predissociation seems to reduce the fluorescence quantum yield to near zero, the states of both CaCCCH3 and SrCCCH3 show strong fluorescence. Although relatively remote from the chromophore (the metal atom), the methyl group perturbs the electronic structure sufficiently to shift the low lying vibrational levels of the state out of the predissociative region.
2A1–
2A1 electronic transitions. The vibrational structure in each system is limited to a short progression in the metal–carbon stretching mode. The Ö origin is blue-shifted relative to the monoacetylides of Ca and Sr. Furthermore, in contrast to CaCCH and SrCCH, where predissociation seems to reduce the fluorescence quantum yield to near zero, the states of both CaCCCH3 and SrCCCH3 show strong fluorescence. Although relatively remote from the chromophore (the metal atom), the methyl group perturbs the electronic structure sufficiently to shift the low lying vibrational levels of the state out of the predissociative region.
Introduction
The monoacetylides of alkaline earth metals have recently attracted considerable interest. These free radicals were first observed by Bopegedera et al. in 1987 using laser-induced fluorescence (LIF) spectroscopy.1 CaCCH and SrCCH were prepared by reaction of metal vapour with acetylene in a Broida oven. Relatively low resolution spectra were obtained showing spin–orbit splittings and some vibrational structure, from which it was deduced that CaCCH and SrCCH are linear in both their ground (
2Σ+) and first excited (Ã
2Π) electronic states. Subsequently, Bopegedera et al., followed by a number of other groups, reported rotationally-resolved LIF spectra of CaCCH.2–6 In addition, Ziurys and co-workers have obtained pure rotational spectra of the
2Σ+ state of CaCCH and SrCCH using millimetre/submillimetre absorption spectroscopy.7,8 A variety of properties, particularly for CaCCH, have now been firmly established including the structure, vibrational frequencies of several modes, spin–rotation and lambda-doubling parameters, and the electric dipole moments in the Ã
2Π1/2 and
2Σ+ states.
MgCCH is another metal acetylide which has generated interest. Stimulated by the possibility of observing this molecule in interstellar space, the first spectroscopic data on MgCCH, obtained using millimetre absorption spectroscopy, was reported by Anderson and Ziurys.9 Shortly afterwards the first LIF spectra of MgCCH were recorded in our laboratory.10,11 The study by Anderson and Ziurys showed that MgCCH is linear in its ground electronic state (2Σ+) while our LIF data showed that linearity is maintained in the first excited state (2Π). One of the interesting features of the alkaline earth monoacetylides is the scope they offer for characterising the Renner–Teller effect in tetra-atomic molecules. Indeed, several detailed studies of the Renner–Teller effect in CaCCH have recently appeared in the literature.12–14 Other published work on MgCCH includes a matrix isolation IR study by Thompson and Andrews15 and high level ab initio calculations by Woon.16,17
In this paper we present the first LIF spectra of the monomethylacetylides of calcium and strontium. In their initial work on the monoacetylides, Bopegedera and co-workers also attempted to obtain LIF spectra of the metal methylacetylide free radicals using methylacetylene instead of acetylene as the reagent.1 They could find no evidence for formation of CaCCCH3 but did observe spectral features which may have been due to SrCCCH3, although these spectra were not published. The signal apparently due to SrCCCH3 was weak and obscured by SrOH bands. We have been able to obtain rather strong vibrationally-resolved LIF spectra of both CaCCCH3 and SrCCCH3 using a laser ablation source combined with supersonic expansion. The cooling in the jet is particularly important as it allows vibronic bands of other species, particularly the metal hydroxides, to be distinguished from the methylacetylides. We have observed the Ã
2E–
2A1 and
2A1–
2A1 transitions of CaCCCH3 and SrCCCH3 and report our findings here.
Experimental
The experimental apparatus and procedure have been described in detail elsewhere.11 Briefly, the metal methylacetylides were produced in a pulsed supersonic expansion using excimer laser (193 nm) ablation of a calcium or strontium target in the presence of a suitable volatile organic precursor. Spectra were obtained by crossing the jet with the beam from a tunable pulsed dye laser (linewidth 0.2 cm−1) approximately 4 cm downstream of the nozzle exit. Excitation spectra were recorded by detection of the unfiltered fluorescence gathered by a f/1.3 lens using a photomultiplier tube (PMT), while dispersed fluorescence spectra were obtained using a 0.27 m monochromator (Acton Research) also equipped with a PMT detector. For both excitation and dispersed fluorescence spectra, the PMT output was digitised and then transferred to a PC on a shot-to-shot basis for gated integration and averaging.The precursor was seeded into helium carrier gas by flowing helium through a temperature-controlled sample bomb. A variety of organic precursors successfully yielded the desired species, including toluene, hexane and benzene. Methylacetylene was also employed in some experiments but no advantage accrued over the liquid precursors. Since the latter were easier to handle, most of the work was carried out with benzene (99.99% purity).
Computational details
To assist in the assignment of vibrational structure, calculations were carried out on several free radicals composed of a calcium atom and a hydrocarbon ligand. DFT calculations using the B3LYP functional were carried out using MOLPRO.18,19 B3LYP was chosen because it has been widely characterised and is known to be superior to the Hartree–Fock method for the prediction of vibrational frequencies.20,21The triple zeta basis sets developed by Schäfer et al. were selected for these calculations.22 For calcium, the basis set chosen was that optimised for the 3P excited state of calcium rather than the 1S ground state. This gives a better description of the valence p space, something which is known to be important for molecules containing an alkaline earth atom. Three d polarisation functions were added with exponents of 0.2, 0.15, and 0.05. For carbon the triple zeta basis set was supplemented with two diffuse p functions (exponents 0.035 and 0.012) to help describe the negatively charged C atom closest to the metal atom, and two d polarisation functions with exponents 0.4 and 1.6. For hydrogen, two p polarisation functions with exponents of 0.4 and 1.6 were added.
Results and discussion
A. Calcium
Laser ablation of metals in the presence of an organic precursor usually produces a variety of gas phase species. For example, during experiments using a calcium ablation target and benzene as the added precursor, we have observed LIF spectra of a number of well-known calcium-containing molecules. CaH, CaCH3, CaCCH, and CaOH were all easily identified under these conditions. CaOH was always seen despite efforts to minimise water contamination in the carrier gas/precursor mixture and elsewhere in the vacuum system.However during the same experiments, which were being carried out to optimise the nozzle conditions for radical production, the LIF spectrum of a previously unidentified species was also observed. Fig. 1 shows a laser excitation scan of the relevant spectral region. In addition to bands due to CaOH and CaCH3, several new bands are present in this spectrum. Any variation of experimental conditions, including using the alternative organic precursors toluene or hexane, produced no change in the relative intensities of these new bands, thus showing that they arise from the same molecule. The spectral carrier is undoubtedly a calcium-containing free radical for a number of reasons including:
(i) The spectrum disappears when another metal is substituted for the calcium target.
(ii) The bands are very close to the Ö transitions of a wide variety of calcium-containing free radicals,23 including CaCCH.1,2
(iii) A spin–orbit doubling of ∼70 cm−1 (see below) is seen in the first excited electronic state which is typical of that found for orbitally degenerate first excited states of calcium-containing free radicals.23
 | ||
| Fig. 1 Excitation spectrum showing the Ö and – regions of CaCCCH3. Vibrational mode ν5 is the Ca–C stretch. This spectrum was recorded with benzene as precursor seeded in helium at a total stagnation pressure of 10 bar. In addition to bands due to CaCCCH3, contributions from CaOH (*) and CaCH3
(+) can also be identified in the spectrum. | ||
However, although the carrier is undoubtedly a free radical of type CaR, the identity of the substituent R is not immediately apparent. Rotational structure would be of considerable assistance but could not be resolved in these experiments. Isotope substitution is also potentially valuable, and some experiments were carried out with benzene-d6 as precursor. However, these did not prove to be definitive. Despite using pure benzene-d6, the extent of deuteration of the spectral carrier was small. Weak shoulders on the low frequency side of certain bands (the 510 bands; see later) in the excitation spectra were observed, indicating the presence of hydrogen and a small isotope shift. However, this does not allow a firm identification of the spectral carrier, since many conceivable Ca–R radicals would show the same behaviour. The reason for the ineffective deuteration is unknown, but it may be related to the abundant CaOH signal seen in these experiments (see below). In any event, the absence of firm evidence from rotational structure and isotope substitution experiments meant that alternative arguments had to be employed to identify the spectral carrier. Fortunately, as detailed below, there is other evidence that helps in the assignment of R.
The origin transition of the first electronic band system consists of a pair of intense bands at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 103 and 16172 cm−1. From their separations and relative intensities, these resemble the spin–orbit doublets commonly found in Ö systems of calcium-containing free radicals. Confirmation is provided by a second, weaker, pair of bands at 16471 and 16542 cm−1, which share the same spin–orbit splitting and are attributable to vibrational excitation in the à state (see later). The importance of the spin–orbit splitting is that it shows that the spectral carrier has an equilibrium geometry with a symmetry sufficiently high for electronic degeneracies to occur (or alternatively that the symmetry-lowering parts of the molecule are too distant from the orbital in which the unpaired electron resides to resolve the orbital degeneracy). This symmetry constraint severely restricts the number of possible spectral carriers.
103 and 16172 cm−1. From their separations and relative intensities, these resemble the spin–orbit doublets commonly found in Ö systems of calcium-containing free radicals. Confirmation is provided by a second, weaker, pair of bands at 16471 and 16542 cm−1, which share the same spin–orbit splitting and are attributable to vibrational excitation in the à state (see later). The importance of the spin–orbit splitting is that it shows that the spectral carrier has an equilibrium geometry with a symmetry sufficiently high for electronic degeneracies to occur (or alternatively that the symmetry-lowering parts of the molecule are too distant from the orbital in which the unpaired electron resides to resolve the orbital degeneracy). This symmetry constraint severely restricts the number of possible spectral carriers.
Having eliminated closed-shell molecules, and recognising the need for a high local symmetry in the vicinity of the calcium atom, it is now possible to identify likely substituents. The observation of strong bands of CaCCH and CaCH3 alongside the new CaR species is an important clue. The likelihood is that the substituent R is a hydrocarbon fragment, particularly given that benzene was used as the precursor for the spectrum in Fig. 1. Although CaOH contributes to the LIF spectrum, and therefore an alternative substituent containing one or more O atoms must also be considered, the evidence against this is strong. CaOH presumably forms either from direct laser ablation of Ca(OH)2 from the surface of the ablation target, or by gas-phase reaction of Ca atoms with H2O or OH (the latter will be formed in the laser ablation plasma). Water vapour leads to very efficient mopping up of calcium atoms. Even when small leaks were present in our gas delivery manifold the spectra were almost completely dominated by CaOH. Spectra such as that shown in Fig. 1 were difficult to obtain and required considerable effort to minimise air leaks and to remove adsorbed water from gas pipes. When such conditions were achieved, there would be little opportunity for other oxygen-containing ligands to form in the laser ablation plasma. This is borne out by the lack of observation of LIF signals of calcium alkoxy radicals,24 including CaOCH3, under our experimental conditions. The evidence is therefore overwhelmingly in favour of a hydrocarbon substituent R.
If CCH and CH3 can be formed during the laser ablation process, then more complex ligands, such as CCCCH, CCCH3, or C(CH3)3 could conceivably be present. The corresponding CaR radicals would all be expected to possess or exceed the minimum three-fold rotational symmetry required for doubly degenerate electronic states. The arguments made so far do not allow us to distinguish between these possibilities, but when the vibrational structure is investigated more closely, as is done in the next section, CaCCCH3 becomes the only feasible carrier.
![[X with combining tilde]](https://www.rsc.org/images/entities/b_char_0058_0303.gif) system.
The prominent bands at 16101 and 16172 cm−1 in Fig. 1 are the lowest wavenumber features observed for the new calcium-containing free radical. As mentioned earlier, these clearly constitute a spin–orbit doublet and, assuming the carrier is CaCCCH3, can be assigned to the Ã
2E1/2–
2A1 and Ã
2E3/2–
2A1 000 transitions, respectively. The Ö transitions in CaR free radicals involve an electron of mainly non-bonding character moving between metal-localised orbitals.23,25 As a result, extensive vibrational structure is not expected in the Ö system and the only mode likely to yield any substantial structure is the Ca–R stretch, since this is the mode which is most strongly perturbed by the electronic transition.
system.
The prominent bands at 16101 and 16172 cm−1 in Fig. 1 are the lowest wavenumber features observed for the new calcium-containing free radical. As mentioned earlier, these clearly constitute a spin–orbit doublet and, assuming the carrier is CaCCCH3, can be assigned to the Ã
2E1/2–
2A1 and Ã
2E3/2–
2A1 000 transitions, respectively. The Ö transitions in CaR free radicals involve an electron of mainly non-bonding character moving between metal-localised orbitals.23,25 As a result, extensive vibrational structure is not expected in the Ö system and the only mode likely to yield any substantial structure is the Ca–R stretch, since this is the mode which is most strongly perturbed by the electronic transition.
Consequently, the assignment of the doublet centred at 16507 cm−1 is to excitation of the Ca–R stretching vibration in the à state, which gives a frequency of ∼370 cm−1 for this mode. To confirm that this doublet is due to excitation of νCa–R in the à state, we have also recorded dispersed fluorescence spectra. The dispersed fluorescence spectrum is essentially the same, apart from a wavelength shift, if the laser is tuned to either the 16471 or 16542 cm−1 transition, as would be expected if they constitute a spin–orbit doublet. The strongest feature is a band 364 cm−1 to the red of the laser wavelength (see Fig. 2). The intensity of this band and its position clearly shows that it is due to single quantum excitation of the Ca–C stretch. We therefore determine νCa–C to be 364 and 370 cm−1 in the ground and first excited electronic states, respectively. The small change in vibrational frequencies between the à and states is consistent with the idea that a non-bonding electron is involved in the electronic transition.
 | ||
| Fig. 2 Dispersed fluorescence spectrum of CaCCCH3 obtained by laser pumping the 16537 cm−1 transition (Ã
2E3/2–
2A1 510 band). The strongest band corresponds to single quantum excitation of the Ca–C stretch (ν5) in the ground electronic state. | ||
Having measured the Ca–R stretching frequencies, this information can now be used to identify R. To put this on a firmer footing, density functional theory (DFT) calculations were carried out. Attention was restricted to the calcium acetylides CaCCH, CaCCCH3, and CaCCCCH. The last two molecules were chosen because they were viewed as the most likely candidates for the spectral carrier. CaCCH was also included because the Ca–C stretching frequency in the ground electronic state of this molecule is known, and therefore serves as a useful reference point.
Given that the bonding in CaR radicals in their ground and low-lying excited electronic states is primarily ionic,23,25 the identity of R is not expected to have a major effect on the strength of the Ca–R bond, especially when the ligands are from the same family (as the acetylides considered above are). In other words, the Ca–R bond force constant will show minor variations with ligand identity, but the Ca–R stretching frequency will be strongly dependent on the effective reduced mass. The similarity in binding is confirmed by the DFT calculations, which predict only small variations in Ca–C equilibrium bond lengths (see Table 1) and diagonal Ca–C stretching force constants (not shown). Consequently, ν(Ca–R) will be largely determined by the mass of substituent R.
| CaCCH | CaCCCH3c | CaCCCCH | |
|---|---|---|---|
| a The B3LYP functional was employed. See text for further details. b The DFT and experimental data refer to the ground electronic states of these molecules. The full set of vibrational frequencies for each molecule is available from the authors on request. c The calculated CCH bond angle was 111.56°. | |||
| r Ca–C/Å | 2.419 | 2.408 | 2.426 |
| r C1–C2/Å | 1.220 | 1.223 | 1.229 |
| r C2–C3/Å | — | 1.461 | 1.365 |
| r C3–C4/Å | — | 1.208 | |
| r C–H/Å | 1.064 | 1.092 | 1.065 |
| ν(Ca–C)/cm−1 | 375 | 325 | 298 |
Given the similarities of these molecules, the DFT calculations should systematically underestimate or overestimate the Ca–C stretching frequencies. We can therefore take the known Ca–C fundamental frequency of CaCCH and employ it as a scaling parameter for the DFT results to determine the frequency of the same mode in CaCCCCH and CaCCCH3. In other words, we place no reliance on the absolute frequencies from the DFT calculations, but instead use them to deduce the differences in the Ca–R stretching frequencies from one acetylide to another.
Table 1 summarises the calculated vibrational frequencies. The DFT calculations underestimate the experimental (fundamental) Ca–C stretching frequency in CaCCH (399 cm−1)1 by approximately 6%. Scaling the frequencies of the other molecules by the same proportion gives predictions of ca. 346 and 317 cm−1 for CaCCCH3 and CaCCCCH, respectively. The latter value is far smaller than the observed ground state fundamental vibrational frequency in this work (364 cm−1) and eliminates CaCCCCH as the spectral carrier. On the other hand, the discrepancy for CaCCCH3 is only 18 cm−1 and could easily be accounted for by limitations in the DFT calculations and the scaling procedure. It should also be borne in mind that the precision in measuring the vibrational frequency from the dispersed fluorescence spectrum is estimated as ±5 cm−1. When all of these factors are taken into account, CaCCCH3 is a plausible candidate for the spectral carrier.
Although other hydrocarbon substituents with similar masses are conceivable, none satisfy the additional requirement of high symmetry necessary to generate spin-orbit splitting. Consequently, the new electronic system is assigned to the Ã
2E–
2A1 transition of CaCCCH3.
![[B with combining tilde]](https://www.rsc.org/images/entities/b_char_0042_0303.gif) – system.
The excitation spectrum of CaCCCH3 shows another strong band at 16861 cm−1. Unlike the Ö system, there is no evidence of a spin-orbit partner and therefore, by analogy with other CaR radicals, the only reasonable assignment is to the
2A1–
2A1 000 transition. A weaker band is also observed at 17223 cm−1 which, like the origin, shows no spin-orbit doubling. This band is 360 cm−1 from the origin, very similar to the Ca–C stretching frequency in the Ã
2E state. We therefore assign this 360 cm−1 interval to excitation of the Ca–C stretch in the
2A1 state.
– system.
The excitation spectrum of CaCCCH3 shows another strong band at 16861 cm−1. Unlike the Ö system, there is no evidence of a spin-orbit partner and therefore, by analogy with other CaR radicals, the only reasonable assignment is to the
2A1–
2A1 000 transition. A weaker band is also observed at 17223 cm−1 which, like the origin, shows no spin-orbit doubling. This band is 360 cm−1 from the origin, very similar to the Ca–C stretching frequency in the Ã
2E state. We therefore assign this 360 cm−1 interval to excitation of the Ca–C stretch in the
2A1 state.
Although the – transitions of many calcium-containing free radicals have been reported, the
2Σ+–
2Σ+ system of CaCCH has not been observed by LIF spectroscopy. From their rotational analysis of the Ö system,2 Bopegedera et al. argued that the absence of an observable
2Σ+ state is most likely due to predissociation, a conclusion drawn from the Λ-doubling parameters determined for the Ã
2Π state. More recent work by Li and Coxon has revealed errors in the analysis of Bopegedera et al.5 In particular, the pν
Λ-doubling parameter was found to be positive, rather than negative as reported by Bopegedera et al. The new value for pν gave an estimated
2Σ+–Ã
2Π separation of 1080 cm−1 in the pure precession limit. This value, which is reasonable when compared with other CaR molecules,23 supports the proposition that predissociation is responsible for the non-observation of LIF spectra for the
2Σ+ state of CaCCH.
The –Ã separation in CaCCCH3 is found to be 758 cm−1. The substitution of a methyl group in place of the hydrogen atom in CaCCH is unlikely to have a major impact on the calcium-localised orbitals. However, its effect clearly cannot be negligible since the state of CaCCCH3 has a substantial fluorescence quantum yield, unlike that of CaCCH. The methyl group presumably decreases the predissociation rate by altering the relative energies of the state and the potentially predissociating state, pushing the latter above the former.
Table 2 summarises the positions and assignments of all CaCCCH3 bands observed in this work.
| Band centre/cm−1 | Assignment |
|---|---|
| 16103 |
Ã
2E1/2–
2A1 000 |
| 16172 |
Ã
2E3/2–
2A1 000 |
| 16471 |
Ã
2E1/2–
2A1 510 |
| 16542 |
Ã
2E3/2–
2A1 510 |
| 16861 |
2A1–
2A1 000 |
| 17223 |
2A1–
2A1 000 |
B. Strontium
The observations with strontium as ablation target are qualitatively similar to calcium. Fig. 3 shows an excitation spectrum obtained with benzene as the precursor. New bands attributable to SrCCCH3 can be seen in addition to contributions from SrCCH and SrOH. The positions of the SrCCCH3 bands are listed in Table 3. The à 2E state of SrCCCH3 is expected to exhibit a larger spin–orbit splitting than that of CaCCCH3 due to the presence of the heavier metal atom in the former molecule. The pair of strong bands at 14831 and 15128 cm−1 are therefore assigned to the Ã
2E1/2–
2A1 and Ã
2E3/2–
2A1 000 transitions of SrCCCH3, respectively. This gives a value of 297 cm−1 for the spin–orbit coupling constant in the Ã
2E state of SrCCCH3, which is clearly much larger than that observed for CaCCCH3 but is quite similar to that reported for the corresponding state in SrCCH (275 cm−1).1
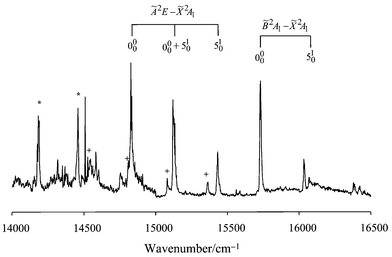 | ||
| Fig. 3 Laser excitation spectrum showing the Ö and – systems of SrCCCH3. Contributions from SrOH and SrCH3 are marked * and + on the spectrum, respectively. Two laser dyes were used to scan the range shown, the changeover being evident from the discontinuity in the baseline at 15000 cm−1. | ||
By analogy with CaCCCH3, some activity in the Sr–C stretching mode is expected in the LIF spectrum of SrCCCH3. The substitution of Sr for Ca will substantially lower the frequency of this vibration. A candidate is the band at 15435 cm−1, which is ∼307 cm−1 from the Ã
2E3/2–
2A1 000 band. Notice that this value is similar to the spin–orbit splitting in the origin band. It is therefore likely that the band due to single quantum excitation of νSr–C in the Ã
2E1/2–
2A1 system overlaps the Ã
2E3/2–
2A1 000 band, and this is consistent with the broadening of the band at 15128 cm−1.
A prominent
2A1–
2A1 system is also observed for SrCCCH3. The strongest band at 15732 cm−1 is assigned to the 000 transition while that at 16036 cm−1 is due to excitation of one quantum in the Sr–C stretch, giving a value of 304 cm−1 for νSr–C in the
2A1 electronic state.
The most recent work on benzene photodissociation, carried out by Tsai and co-workers using ion imaging,26 has established that there is only one single-photon decomposition process at 193 nm, viz.
| C6H6+hν→C6H6*→C6H5+H | (1) |
Other fragments formed in the photolysis of benzene, which were previously thought to be the result of minor single-photon channels,27 were found by Tsai et al. to be due to two-photon processes proceeding via the ‘hot’ benzene:
| C6H6*+hν→C6H4+H2 | (2) |
| C6H6*+hν→C4H3+C2H3 | (3) |
| C6H6*+hν→C5H3+CH3 | (4) |
Secondary photodissociation of the products of reactions (2)–(4) will yield other fragments. For example, loss of H from C2H3 will generate acetylene, and this is a well-known product from the photolysis of benzene.28 Although this is a minor product under normal low light intensity conditions, under our conditions it is expected to be a major product. Acetylene itself will undergo photodissociation at 193 nm and the resulting reaction
| C2H2+hν→C2H+H | (5) |
Recombination of the products of reactions (4) and (5) will then lead to the formation of methylacetylene, viz.
| C2H+CH3+M→CH3CCH | (6) |
| M*+C2H2→MCCH+H | (7) |
| M*+CH3CCH→MCCCH3+H | (8) |
Conclusions
The first laser-induced fluorescence spectra of the CaCCCH3 and SrCCCH3 free radicals have been reported. The à 2E–
2A1 and
2A1–
2A1 systems have been observed for both molecules. The vibrational structure is sparse, with only short progressions being identified in the metal-carbon stretching mode. The observation of strong – transitions contrasts sharply with CaCCH and SrCCH, where the analogous bands are not observable by LIF spectroscopy. Detailed rotationally-resolved studies would be valuable in shedding light on this apparent anomaly.
Acknowledgements
The authors acknowledge the support of this work by the UK Engineering and Physical Sciences Research Council.References
- A. M. R. P. Bopegedera, C. R. Brazier and P. F. Bernath, Chem. Phys. Lett., 1987, 136, 97 CrossRef CAS.
- A. M. R. P. Bopegedera, C. R. Brazier and P. F. Bernath, J. Mol. Spectrosc., 1988, 129, 268 CrossRef CAS.
- C. J. Whitham, B. Soep, J. P. Visticot and A. Keller, J. Chem. Phys., 1990, 93, 991 CrossRef CAS.
- A. J. Marr, J. Perry and T. C. Steimle, J. Chem. Phys., 1995, 103, 3861 CrossRef CAS.
- M. Li and J. A. Coxon, J. Mol. Spectrosc., 1996, 176, 206 CrossRef CAS.
- M. G. Li and J. A. Coxon, J. Mol. Spectrosc., 1997, 184, 395 CrossRef CAS.
- M. A. Anderson and L. M. Ziurys, Astrophys. J., 1995, 444, L57 CrossRef CAS.
- B. Nuccio, A. J. Apponi and L. M. Ziurys, Chem. Phys. Lett., 1995, 247, 283 CrossRef CAS.
- M. A. Anderson and L. M. Ziurys, Astrophys. J., 1995, 439, L25 CrossRef CAS.
- G. K. Corlett, A. M. Little and A. M. Ellis, Chem. Phys. Lett., 1996, 249, 53 CrossRef CAS.
- G. K. Corlett, M. S. Beardah and A. M. Ellis, J. Mol. Spectrosc., 1997, 185, 202 CrossRef CAS.
- M. G. Li and J. A. Coxon, J. Mol. Spectrosc., 1996, 180, 287 CrossRef CAS.
- M. G. Li and J. A. Coxon, J. Mol. Spectrosc., 1997, 183, 250 CrossRef CAS.
- M. G. Li and J. A. Coxon, J. Mol. Spectrosc., 1999, 196, 14 CrossRef.
- C. A. Thompson and L. Andrews, J. Am. Chem. Soc., 1996, 118, 10242 CrossRef CAS.
- D. E. Woon, Astrophys. J., 1996, 456, 602 CrossRef CAS.
- D. E. Woon, Chem. Phys. Lett., 1997, 274, 299 CrossRef CAS.
- MOLPRO, a package of ab initio programs designed by H-J. Werner and P. J. Knowles, version 2002.1, R. D. Amos, A. Bernhardsson, A. Berning, P. Celani, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, C. Hampel, G. Hetzer, P. J. Knowles, T. Korona, R. Lindh, A. W. Lloyd, S. J. McNicholas, F. R. Manby, W. Meyer, M. E. Mura, A. Nicklass, P. Palmieri, R. Pitzer, G. Rauhut, M. Schütz, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and H-J. Werner.
- G. Rauhut, A. El Azhary, F. Eckert, U. Schumann and H.-J. Werner, Spectrochim. Acta, 1999, 55, 651 Search PubMed.
- G. Rauhut and P. Pulay, J. Phys. Chem., 1995, 99, 3093 CrossRef CAS.
- A. P. Scott and L. Radom, J. Phys. Chem., 1996, 100, 16502 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef.
- P. F. Bernath, in Advances in Photochemistry, ed. D. C. Neckers, D. H. Volman and G von Bünau, Wiley, 1997, vol. 23, pp. 1–62 Search PubMed.
- C. R. Brazier, L. C. Ellingboe, S. Kinsey-Nielson and P. F. Bernath, J. Am. Chem. Soc., 1986, 108, 2126 CrossRef CAS.
- A. M. Ellis, Int. Rev. Phys. Chem., 2001, 20, 551 CrossRef CAS.
- S.-T. Tsai, C.-L. Huang, Y. T. Lee and C.-K. Ni, J. Chem. Phys., 2001, 115, 2449 CrossRef CAS.
- A. Yokoyama, X. Zhao, E. J. Hintsa, R. E. Continetti and Y. T. Lee, J. Chem. Phys., 1990, 92, 4222 CrossRef CAS.
- J. K. Foote, M. H. Mallon and J. N. Pitts, J. Am. Chem. Soc., 1966, 88, 3698 CrossRef CAS.
- A. Läuter, K. S. Lee, K. H. Lung, R. K. Vatsa, J. P. Mittal and H.-R. Volpp, Chem. Phys. Lett., 2002, 358, 314 CrossRef CAS.
- V. S. Burakov, N. V. Tarasenko and N. A. Savastenko, Spectrochim. Acta, Part B, 2001, 56, 961 CrossRef.
- T. J. Hughes and M. R. Levy, Phys. Chem. Chem. Phys., 2000, 2, 651 RSC.
| This journal is © the Owner Societies 2003 |