Matrix-isolation FT-IR spectra and molecular orbital calculations on neutral N,N-dimethylglycine†
A. Gómez-Zavagliaab, I. D. Revaa and R. Fausto*a
aDepartment of Chemistry, University of Coimbra, P-3004-535, Portugal
bFacultad de Farmacia y Bioquímica, Universidad de Buenos Aires, RA-1113, Argentina. E-mail: rfausto@ci.uc.pt
First published on 27th November 2002
Abstract
The structures and vibrational spectra of the preferred conformers of the neutral form of N,N-dimethylglycine (DMG) were studied by a combined approach, using DFT(B3LYP)/6-311++G** and MP2/6-31++G** calculations and low temperature matrix isolation IR spectroscopy. The conformational ground state was found to be the intramolecularly O–H⋯N hydrogen-bonded GAT form, where the (lone pair)-N–C–C and N–C–C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O dihedral angles are 30°
(gauche; G) and ca. 180°
(anti; A), respectively, and the carboxylic group assumes the trans
(T) configuration (OC–O–H dihedral angle equal to 180°). The presence in the matrices of two additional conformers, where the carboxylic moiety assumes the most commonly found cis
(C) conformation and the N–C–CO axis adopts the syn arrangement (the two conformers differ only in the positions of the methyl groups), could also be established. Observation of these conformers is in consonance with the theoretical predictions, which indicate that the observed conformers should differ in energy by less than 7 kJ mol−1. Full assignment of the observed infrared spectra of both DMG and its -OD isotopomer in Ar and Xe matrices was carried out on the basis of comparison with the theoretically predicted spectra and temperature variation experiments.
O dihedral angles are 30°
(gauche; G) and ca. 180°
(anti; A), respectively, and the carboxylic group assumes the trans
(T) configuration (OC–O–H dihedral angle equal to 180°). The presence in the matrices of two additional conformers, where the carboxylic moiety assumes the most commonly found cis
(C) conformation and the N–C–CO axis adopts the syn arrangement (the two conformers differ only in the positions of the methyl groups), could also be established. Observation of these conformers is in consonance with the theoretical predictions, which indicate that the observed conformers should differ in energy by less than 7 kJ mol−1. Full assignment of the observed infrared spectra of both DMG and its -OD isotopomer in Ar and Xe matrices was carried out on the basis of comparison with the theoretically predicted spectra and temperature variation experiments.
Introduction
N,N-dimethylglycine (DMG) is present in living cells, as a product of the metabolic pathways of choline and aminoacids. Choline is a compound present in the membrane of the eucariotic cells, and its catabolism in mitochondria is a cyclical process, involving the formation of betaine, DMG, sarcosine, glycine and serine, with the ultimate regeneration of choline. The deficiency of dimethylglycine dehydrogenase, one of the enzymes involved in this metabolic pathway, may produce an increase of the serum and urine levels of DMG, which is translated as a chronic muscle fatigue among other symptoms.1 Metabolism of aminoacids involves several enzymes, and the deficiency of some of them may also lead to the accumulation of DMG in serum and urine. Several methods have been described for the quantification of this metabolite either in serum or in urine.2DMG is found in very low levels in foods such as cereal grains, seeds and meats. Since it is a sweet-tasting substance legally considered as a nutrient, it is also used for supplementation of diet tablets and “energetic drinks” for sportsmen.3 It is an anti-stress nutrient with antioxidant properties. Recently, studies have implicated the generation of oxygen-derived free radicals and lipid peroxidation as one of the mechanisms in the pathogenesis of gastric ulcer. It has been reported that the use of DMG in these cases displays protective effects due to its free radical scavenging activity and the cytoprotection of gastric mucosa.4 In addition, DMG was also found to give rise to immunomodulatory effects in cats fed with DMG-supplemented diets.5
From a chemical point of view, DMG belongs to the family of N-methylated derivatives of glycine, which also includes sarcosine (N-methylglycine) and betaine (N,N,N-trimethylglycine) as members. The later compounds are also well known by their biochemical relevance.6–8 In spite of their biological importance, the fundamental information available on the structure and spectroscopic properties of these molecules is relatively scarce, in particular in the case of DMG.
Theoretical calculations have been used successfully to analyse relative energies and geometries of the conformers of a fairly large number of aminoacids (see, for example, refs. 9 and 10 and references therein). Concerning DMG, results of a theoretical study were reported by Headley and Starnes.11 In that study, ab initio calculations undertaken at both the Hartree–Fock and single point second order Møller–Plesset levels of theory (with the 6-311++G** basis set) were used to predict the structures and relative energies of the most stable conformers of DMG. Five conformers were predicted by the Hartree–Fock calculations, with the most stable form exhibiting an OH⋯N intramolecular hydrogen bond and the carbonyl group in the trans orientation (OC–O–H angle equal to 180°). This result contrasted with the available data for glycine, where the most stable conformation has the carboxylic moiety in the cis configuration (OC–O–H equal to 0°) and is stabilized by intramolecular N–H⋯O hydrogen bonds.9
In a more recent publication, Headley and Starnes examined the gas phase tautomerization of DMG from its neutral form to the zwitterionic species, which was predicted to be higher in energy than the ground state species by more than 40 kJ mol−1.12
To the best of our knowledge, besides the studies of Headley et al. further structural studies on DMG have not been published. In particular, the X-ray structure of this compound has not yet been published. Moreover, no vibrational data has been reported until now.
Since the characterization of the most stable conformers of DMG and the factors that contribute to their relative stability is essential to fully understand its biological properties, in this work we used the density functional theory (DFT) and Møller–Plesset second order (MP2) methods in order to perform the conformational analysis of monomeric neutral DMG and predict the infrared spectra of their relevant conformers. These results were then used to interpret the observed spectra for the studied molecule (including its -OD substituted isotopomer, DMG-OD) isolated in argon and xenon matrices.
Materials and methods
Computational methodology
The quantum chemical calculations were performed with Gaussian 9813 at the DFT and MP2 levels of theory, using the 6-311++G** and 6-31++G** basis sets, respectively.14 Conformations were optimized at each level of theory using the geometry direct inversion of the invariant subspace (GDIIS) method.15 Vibrational frequencies were calculated at each level of theory and the nature of the critical points on the potential energy surface resulting from optimisation was determined by inspection of the corresponding calculated Hessian matrix, all structures confirmed to be minimum energy conformations. The calculated frequencies were scaled down by a single factor (0.978) to correct them for the effects of basis set limitations, neglected part of electron correlation and anharmonicity effects, and used to assist the analysis of the experimental spectra and to account for the zero-point vibrational energy contribution to the total energy. Normal coordinates analyses were undertaken in the internal coordinates space as described by Schachtschneider16 using the program BALGA and the optimised geometries and harmonic force constants resulting from the DFT/B3LYP calculations. Potential energy profiles for internal rotation were calculated performing a relaxed scan on the potential energy surface (PES) along the reaction coordinate and the transition state structures for conformational interconversion obtained using the synchronous transit-guided quasi-newton (STQN) method.17Matrix isolation infrared spectroscopy
N,N-dimethylglycine was obtained from Aldrich (purity 99%). In the matrix isolation experiments, a glass vacuum system and standard manometric procedures were used to deposit the matrix gas (argon, 99.99990%; xenon, 99.995%, obtained from Air Liquid). Matrices were prepared by co-deposition onto a cooled CsI substrate of the matrix gas and DMG placed in a specially designed temperature variable mini-oven assembled inside the cryostat. The temperature of the mini-oven used to evaporate DMG was, in all experiments, ca. 323 K. At this temperature sublimation occurs without degradation as noticed by the absence of any band in the spectrum that could be ascribed to products of decomposition (e.g., CO2, CO). To check the thermal stability of the compound, we also performed differential scanning callorimetry essays, which showed that decomposition of DMG starts only after melting (above ca. 353 K). Data collection was performed with 0.5 cm−1 spectral resolution on a Mattson (Infinity 60AR Series) FTIR spectrometer. All experiments were done on the basis of an APD Cryogenics close-cycle helium refrigeration system with a DE-202A expander. Necessary modifications of the sample compartment of the spectrometer were made in order to accommodate the cryostat head and allow efficient purging of the instrument by a stream of dry air to remove water and CO2 vapours. After depositing the compound, annealing experiments were performed up to a temperature of 25 K and 60 K for Ar and Xe, respectively.Deuteration of DMG was carried out as described previously18 by multiple recrystallization of commercial DMG from cyclohexane in the presence of D2O in a relation of 1 mol DMG mol−1 D2O. Since DMG has only one labile proton, after deuteration the compound can adopt only pure OD or pure OH form (in the case of partial OH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) →OD exchange). This detail is very important for further analysis because the pure deuterated spectrum can be easily obtained by subtraction of the spectrum of the OH form “impurity” from that of the deuterated sample. The degree of deuteration obtained was higher than 95%.
→OD exchange). This detail is very important for further analysis because the pure deuterated spectrum can be easily obtained by subtraction of the spectrum of the OH form “impurity” from that of the deuterated sample. The degree of deuteration obtained was higher than 95%.
Results and discussion
Molecular geometries and energies
DMG has three different internal rotation axes that can give rise to conformational isomers (Lp-N–C–C, N–C–CO and OC–O–H; Lp=lone electron pair of nitrogen atom).It is well known that, unless a specific intramolecular interaction stabilizes a different conformation, the carboxylic moiety assumes preferentially a planar geometry, with the cis configuration (OC–O–H angle equal to 0°) corresponding to the most stable orientation.19–21 The usual energy difference between the trans and the cis configurations of the OC–O–H axis in carboxylic acids amounts to ca. 15–20 kJ mol−1.19–21 This makes experimental observation of trans-like conformers relatively rare. However, the presence of intramolecular hydrogen bonding has important implications regarding preferred molecular structures. When the hydroxyl carboxylic group participates in an intramolecular hydrogen bond with a neighbouring acceptor group, conformers with a trans OC–O–H axis may be strongly stabilized and assume practical importance.19,21 In DMG, stabilization of carboxylic trans-like conformations by intramolecular OH⋯N bonding is possible, thus imposing the detailed study of the DMG potential energy surface region corresponding to this type of structure in addition to the region associated with the cis carboxylic moiety.
The general preferred conformations of the remaining two internal rotational axis exhibited by DMG are not easily foreseen as well, since the energy differences associated with them can be expected to be relatively small.9,22,23
Hence, in this study, a systematic search on the potential energy surface of DMG was performed at the DFT(B3LYP)/6-311++G** level of theory. The number of structures considered to optimisation was 32, corresponding to all possible relevant combinations of the OC–O–H (0° and 180°); N–C–CO (0°, 120°, −120° and 180°) and Lp-N–C–C (0°, 60°, −60° and 180°) starting dihedral angles. Eight different minima (most of them corresponding to doubly-degenerated-by-symmetry states) could be found. The results, which improve on the previous data obtained at the Hartree–Fock level,11,12 are summarized in Table 1 and Fig. 1 as well as in Table S1 and Fig. S1 deposited as Electronic supplementary material (ESI)†.
| Conformer | DFT(B3LYP)/6-311++G** ΔEZPE | MP2/6-31++G** ΔEZPEb |
|---|---|---|
| a Energies in kJ mol−1
(1 Eh=2625.5001 kJ mol−1); conformers are depicted in Fig. 1 and Fig. S1 (ESI)1.b Zero point energy corrections taken from DFT(B3LYP)/6-311++G** calculations.c Total energies with zero point vibrational energy contribution. | ||
| GAT | 0.0 (−953120.08)c | 0.0 (−950128.22)c |
| ASC | 2.73 | 3.84 |
| GSC | 5.44 | 6.72 |
| GSkC | 9.18 | 9.06 |
| AST | 25.35 | 28.22 |
| GST | 28.44 | 32.09 |
| AAT | 29.87 | 35.61 |
| ASkT | 32.41 | 33.06 |
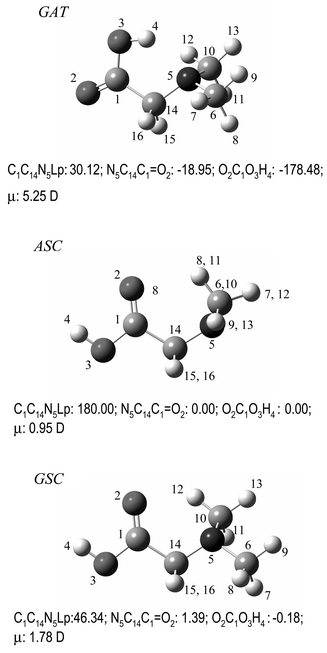 | ||
| Fig. 1 Lowest energy conformers of neutral DMG with atom numbering scheme. For convenience, the values of main dihedral angles (degrees) are included. The whole set of conformers of DMG is shown in Fig. S1 (ESI)†. | ||
Table 1 shows the relative energies of the various DMG minima. Three conformers were predicted to have relative energies within ca. 7 kJ mol−1, then being expected to contribute significantly to the gas-phase conformational equilibrium of DMG: the conformational ground state, GAT, was found to be intramolecularly O–H⋯N hydrogen-bonded, with the Lp-N–C–C and N–C–CO dihedral angles equal to 30°
(gauche; G) and ca. 180°
(anti; A), respectively, and the carboxylic group assuming the trans
(T) configuration; the second and third more stable conformers (ASC and GSC) have a cis
(C) carboxylic group and a syn N–C–CO axis, differing in the conformation around the N–C bond, which is anti
(A) in the second most stable conformer predicted by the calculations and gauche
(G) in the third. The fourth most stable conformer differs from the third one in the conformation of the N–C–CO, which is skew in this case (see Fig. S1 (ESI)†). Its predicted population at room temperature (assuming the Boltzmann distribution) is lower than 1%, so being unimportant in practical terms. The remaining conformers have still substantially higher relative energies (larger than 25 kJ mol−1).
Calculations were performed using both DFT and MP2 methods. Both methods predicted the energy of the conformers in the same order. However, the relative energies to the conformational ground state were calculated systematically larger at the MP2 level. It is generally accepted that the MP2 method is slightly more reliable in estimating energies than the DFT approach (when the same basis set is used), in particular when H-bonding interactions need to be taken into account. This was shown, for instance, in our recent studies on a series of carboxylic acids.24,25 Hence, the higher relative stability of the GAT conformer predicted at the MP2 level of theory seems to be a direct consequence of the better description of the intramolecular OH⋯N hydrogen bond that is the most important factor responsible for the stabilization of this form. This conclusion should, however, been taken with care, since the basis set employed in this study for the MP2 calculations was somewhat smaller than that used for the DFT calculations and, therefore, part of the effect may also be attributed to the expected larger basis set superposition error (BSSE) in the former calculations.
When the structural parameters of the different conformers are compared (see Table S1 (ESI)† and Fig. 2), three clusters are in general observed: the first one includes the cis OC–O–H conformers, which have a relatively low energy; the second, the higher energy trans conformers; the most stable conformer (GAT) appears as an isolated species. The same situation occurs for the electric dipole moments, with the lowest energy conformer having the highest value of this property (Fig. 2a). Larger dipole moments for trans carboxylic conformations have been shown to be a general trend.26 The highest value found for the GAT conformer can be easily rationalized in terms of the relative position of the OH and amino groups in this form and participation of these groups in the intramolecular OH⋯N bonding.
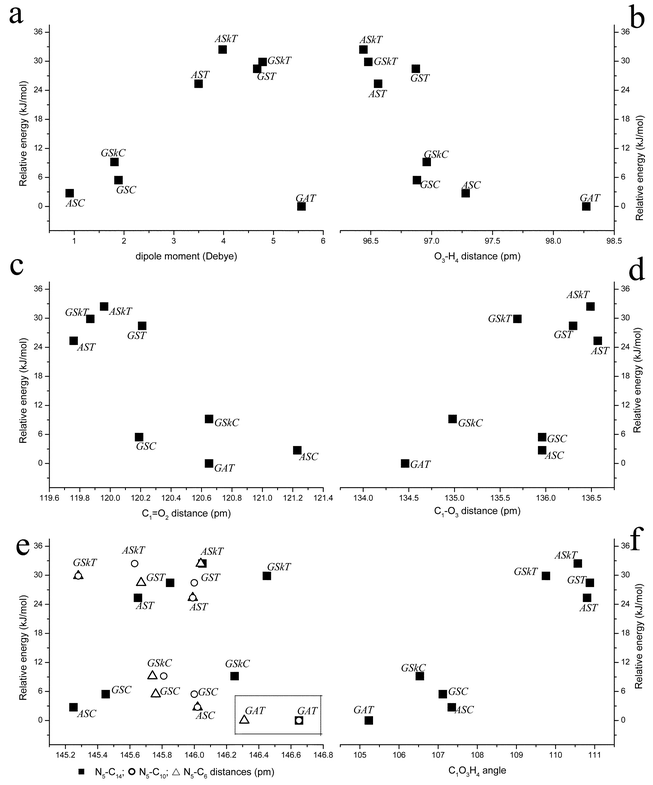 | ||
| Fig. 2 Calculated DFT(B3LYP)/6-311++G** dipole moments and geometrical parameters in DMG conformers. Ordinate positions are defined by the relative energies of the conformers: a: dipole moment; b: O3–H4 bond; c: C1O2; d: C1–O3; e: ■ N5–C14, ○ N5–C10, △ N5–C6; f: C1–O3–H4. | ||
An analysis of the relative values assumed by the structural parameters in the various conformers enables us to establish relevant correlations between these parameters and the relative stability of the conformers.
Several studies concerning the correlation of the O–H bond distance and the relative strength of OH⋯X intramolecular hydrogen bonds (with X=O, N) have been reported, dealing with different families of molecules, including aminoacids and aminoalcohols.27–30 As it could be anticipated, in DMG the O–H distance assumes its largest value in the GAT conformer, leading to a N5⋯H4 distance of 196.64 pm. Taking into account this distance, the enthalpy of the H-bond can be calculated according to Rozenberg et al.30 using the relation −ΔH/kJ mol−1=0.134×(r/nm)−3.05
(with r being the N5⋯H4 distance), yielding an energy for the hydrogen bond in the GAT conformer of −19.1 kJ mol−1. Following the general trend for carboxylic compounds, the O–H bond length in the cis conformers is larger than in the trans forms. This trend was interpreted in detail elsewhere,20 being essentially a consequence of the different alignment of the O–H and CO bond dipole moments in these two conformations. It is also important to point out here that among the cis conformers, the ASC has a longer O–H distance than the GSC, thus being slightly more acidic than this later form (Fig. 2b).
The C–O and CO distances also follow the usual pattern found in carboxylic acids,20 being respectively longer and shorter in trans than in cis conformers as a consequence of the more important –C(O)–OH↔–C(O−)O+H mesomerism within the carboxylic group in the cis configuration (Fig. 2c and d). Note that the largest difference between the bond lengths of the ASC and GSC conformers occurs for CO. Indeed, the ASC conformer displays the longest CO distance among all DMG conformers (121.2 pm), while this bond length in GSC is the shortest within the cis DMG group of conformers, being nearly equal to the CO distance in the trans forms. So, among the low energy cis forms, the GSC conformer is the one having a better hydrogen acceptor carbonyl oxygen atom. As it will be shown later on, this fact (together with the larger acidity associated with the hydroxylic hydrogen atom in this conformer) has important consequences regarding the relative trends for aggregation shown by the experimentally observed conformers.
Concerning the N5–C10, N5–C6 and N5–C14 distances, the calculations indicate that the GAT conformer displays the longest bond lengths. This fact may also be correlated with the existence in this conformer of the intramolecular OH⋯N hydrogen bond, which slightly changes the hybridization state of the nitrogen atom, increasing its sp3 character with the consequent increase in the bond lengths (Fig. 2e).
The C1–C14–N5 and C1–O3–H4 angles also clearly show the presence of the OH⋯N bond in the GAT conformer, assuming their minimum values in this conformer in order to provide a better geometry approach for the establishment of the intramolecular hydrogen bond. As usual,20 in the remaining conformers, the C1–O3–H4 angle is larger when the carboxylic group is trans than when it assumes the cis configuration (Fig. 2f).
Having located all minima on the DMG potential energy surface (PES), an effort was made to locate the conformational transition states connecting the lowest energy minima. The results are summarized in Table 2. The DFT(B3LYP)/6-311++G** calculated energy barrier (from the bottom of the potential wells) between the two equivalent-by-symmetry GAT forms is as small as 2.16 kJ mol−1. Indeed, when zero point vibrational energies are taken into account, this barrier further reduces to 1.44 kJ mol−1. On the other hand, the conversion of both ASC and GSC conformers into the most stable GAT form is associated with energy barriers larger than 50 kJ mol−1, which may be easily overcome in the gaseous phase at room temperature but are large enough to prevent these processes to occur in the low temperature matrices. Fig. S2 (ESI)† presents the calculated potential energy profile for the ASC↔GSC interconversion processes, that correspond essentially to the internal rotation around the N5–C14 bond (though in the transition state structure the OC–C–N dihedral was also found to be considerably deviated from the value it assumes in both the ASC and GSC forms—ca. 0°; see Fig. 1). It shall be noticed that, in spite of the fact that the difference of energy between these two conformers is relatively low (ca. 2.5 kJ mol−1), the energy barriers for both the direct and reverse reaction amount to more than 25 kJ mol−1.
| Transition state structuresb | |||||
|---|---|---|---|---|---|
| TS1 | TS2 | TS3 | TS4 | TS5 | |
| a ΔE correspond to the energy barriers from the bottom of the potential energy minima; the energies of the ASC and GSC conformers relative to GAT are 4.64 and 7.47 kJ mol−1, respectively.b The prime is used to distinguish between the two equivalent-by-symmetry forms of either GAT and GSC conformers. | |||||
| GAT↔GAT′ | GAT→ASC | GAT→GSC | ASC→GSC | GSC↔GSC′ | |
| ΔE | 2.16 | 34.09 | 57.83 | 29.45 | 10.92 |
| ASC→GAT | GSC→GAT | GSC→ASC | |||
| ΔE | 29.45 | 50.36 | 26.74 | ||
Matrix-isolation spectra
The matrix-isolation results obtained in this study are summarized in Tables 3 and S2 (ESI)†, which contain the proposed assignments for the observed bands, and Figs. 3–5. Tables S3–S9 (ESI)† present the results of the vibrational calculations for the experimentally relevant conformers of DMG and DMG-OD.| Approximate description | Conformer | Calculated frequency/cm−1 | ν/cm−1 in argon | ν/cm−1 in xenon |
|---|---|---|---|---|
| a ν, Stretching, δ, bending, γ rocking, tw, twisting, τ torsion, w, wagging, ?, doubtful assignment. See ESI1 for definition of symmetry coordinates, full set of calculated frequencies and intensities and PED. | ||||
| νO–H | GSC | 3680.9 | 3568.2; 3565.9 | 3537.2 |
| ASC | 3663.0 | 3561.6; 3560.4; 3554.8 | 3535.9 | |
| GAT | 3409.9 | 3175.0 | 3174.0 | |
| νCH3as″(1) | GSC | 3061.2 | 3020.6 | 2994.2 |
| GAT | 3042.6 | 3001.2; 2993.0 | 2987.1; 2980.0 | |
| ASC | 3036.9 | 2993.0 | 2980.0 | |
| νCH3as″(2) | GAT | 3039.8 | 3001.2; 2993.0 | 2987.1; 2980.0 |
| ASC | 3034.7 | 2993.0 | 2980.0 | |
| GSC | 3032.3 | 2987.5 | — | |
| νCH2as | ASC | 3009.9 | 2981.5 | 2975.8 |
| GAT | 3007.9 | 2981.5 | 2975.8 | |
| GSC | 2996.2 | 2947.5 | 2939.0 | |
| νCH3as′(1) | GSC | 3007.2 | 2981.2 | 2971.8 |
| GAT | 3003.1 | 2964.2 | 2953.8 | |
| ASC | 2999.8 | 2964.2 | 2953.8 | |
| νCH3as′(2) | ASC | 3000.4 | 2964.2 | 2953.8 |
| GAT | 3000.6 | 2961.7 | 2950.3 | |
| GSC | 2989.4 | 2947.5 | 2939.0 | |
| νCH2s | ASC | 2970.1 | 2925.6 | 2916.4 |
| GAT | 2944.7 | 2914.5; 2904.9; 2900.6 | 2905.7; 2896.2; 2891.5 | |
| GSC | 2832.8 | 2782.8; 2779.0 | 2769.1 | |
| νCH3s(1) | ASC | 2928.1 | 2890.5 (?) | 2878.4 (?) |
| GAT | 2895.0 | 2869.9; 2848.1 | 2864.1; 2839.2 | |
| GSC | 2851.0 | 2808.0; 2802.7 | 2793.5 | |
| νCH3s(2) | ASC | 2922.0 | 2886.7 (?) | 2878.4 (?) |
| GAT | 2890.2 | 2845.4; 2836.8 | 2834.4; 2829.1 | |
| GSC | 2839.9 | 2787.6 | 2774.4 | |
| τC–O+γCH3′(2) | GAT | 1834.1; 1830.0 | 1829.3 | |
| τC–O+νC–N | GAT | 1814.6; 1809.5 | 1807.7 | |
| ASC | 1801.3 | |||
| νCO | GAT | 1803.6 | 1801.9; 1799.0 | 1792.9 |
| GSC | 1788.3 | 1793.1; 1778.4 | 1778.2; 1775.6 | |
| ASC | 1776.0 | 1781.5; 1774.7; 1769.3 | 1780.0; 1772.5; 1769.6 | |
| δCH3as″(2) | ASC | 1497.9 | 1480.2 | 1474.8 |
| GAT | 1467.6 | 1465.1 | 1460.6 | |
| GSC | 1462.9 | 1461.7 | 1456.9 | |
| δCH3as″(1) | GAT | 1481.4 | 1475.2; 1472.0 | 1470.3; 1467.3 |
| ASC | 1483.9 | 1475.2 | 1470.3 | |
| GSC | 1478.5 | 1475.2 | 1470.3 | |
| δCH3as′(1) | GSC | 1473.3 | 1472.0 | 1467.3 |
| GAT | 1469.8 | 1465.1 | 1460.6 | |
| ASC | 1461.9 | 1459.6 | 1455.0 | |
| δCH3s(1) | GAT | 1452.2 | 1451.2; 1447.9 | 1447.9; 1446.6; 1444.0; 1442.3 |
| GSC | 1447.4 | — | — | |
| ASC | 1445.6 | 1434.6 | 1430.4 | |
| δCH3as′(2) | GAT | 1454.8 | 1451.2; 1447.9 | 1447.9; 1446.6; 1444.0 |
| ASC | 1448.8 | 1440.9 | — | |
| GSC | 1454.6 | — | — | |
| δCH2 | GAT | 1429.1 | 1425.2 | 1420.5; 1418.1 |
| GSC | 1422.8 | 1417.5 | 1413.1 | |
| ASC | 1411.9 | 1407.8; 1406.0 | 1401.1 | |
| δCH3s(2) | GSC | 1410.3 | — | — |
| GAT | 1403.9 | 1414.4; 1413.0 | 1410.4 | |
| ASC | 1413.7 | 1407.8; 1406.0 | 1401.1 | |
| δCOH (Fermi resonance with τC–O+γCO) | GAT | 1370.8 | 1387.8; 1346.4; 1344.0 | 1397.2; 1388.2; 1386.2; 1379.1; 1341.4 |
| wCH2 | GSC | 1386.4 | 1376.5 | 1371.0 |
| ASC | 1362.4 | 1359.8 | 1352.2 | |
| GAT | 1328.7 | 1330.7; 1326.1 | 1326.0; 1321.3 | |
| γCH3′(2) | ASC | 1303.8 | 1307.9 | 1310.5 |
| twCH2 | ASC | 1325.3 | 1321.3 | 1323.8 |
| GSC | 1293.7 | 1303.0 | 1306.3 | |
| GAT | 1274.0 | 1280.8; 1277.0 | 1279.5 | |
| γCH3′(1) | GSC | 1281.0 | 1284.8 | 1282.7 |
| GAT | 1269.2 | 1272.8 | 1271.5 | |
| δCOH | GSC | 1268.3 | 1266.5 | — |
| ASC | 1266.6 | 1264.0 | — | |
| 2× δOCO | ASC | 1227.8 | 1221.3 | |
| 2× τCO | ASC | 1213.9; 1212.7 | 1210.0; 1206.0; 1194.0 | |
| νN–Cas | GSC | 1184.6 | 1193.5 | 1190.1; 1188.6 |
| ASC | 1165.2 | 1174.6 | 1174.0 | |
| GAT | 1164.2 | 1171.0 | 1171.4; 1169.6 | |
| νC–O | GAT | 1184.3 | 1190.4 | 1184.7; 1183.7 |
| ASC | 1114.1 | 1117.5; 1114.7 | 1115.1; 1112.9; 1111.6 | |
| GSC | 1104.8 | 1109.9; 1108.0 | 1108.6; 1107.6 | |
| γCH3″(1) | ASC | 1156.4 | 1162.9; 1160.4 | 1157.3; 1156.2 |
| GSC | 1147.1 | 1147.7 | 1147.7; 1144.3 | |
| GAT | 1137.4 | 1141.6; 1139.3 | 1142.1; 1139.4; 1138.5 | |
| γCH3″(2) | GSC | 1093.0 | — | — |
| GAT | 1093.1 | 1097.9 | 1095.3 | |
| ASC | 1092.8 | 1099.0 | 1096.7 | |
| νC–N | ASC | 1063.9 | 1067.0; 1063.1 | 1068.4; 1065.7; 1064.5; 1062.1 |
| GSC | 1051.6 | 1059.5 | 1058.7 | |
| GAT | 1027.3 | 1041.4; 1037.3 | 1043.2; 1041.6; 1040.5; 1038.9; 1035.0 | |
| γCH3′(1) | ASC | 1049.6 | — | 1056.1 |
| γCH3′(2) | GSC | 1039.4 | 1049.6 | 1050.1 |
| GAT | 1035.1 | 1046.3 | 1047.4; 1045.0 | |
| γCH2 | GSC | 975.4 | 985.3 | 981.5 |
| GAT | 966.1 | 980.7 | 977.1 | |
| ASC | 960.3 | 966.9 | 966.1 | |
| δCCO+γCO | ASC | 893.0 | ||
| δC–C | GSC | 872.4 | 890.0 | 886.9 |
| τC–O+τCH3as | ASC | 879.0 | ||
| νC–C | GAT | 856.2 | 872.6 | 874.7 |
| ASC | 815.1 | — | 837.0 | |
| νN–Cs | GAT | 846.7 | 864.2; 862.8; 858.6; 856.8 | 865.8; 863.3; 860.3; 854.9 |
| GSC | 840.9 | 846.3; 837.0 | 856.2 | |
| ASC | 832.9 | 858.7; 853.1 | 858.7; 853.9 | |
| τC–O | GAT | 819.5 | 802.9; 796.9; 787.6; 772.1 | 799.8; 780.2; 755.9 (?) |
| ASC | 640.3 | 619.8; 510.5 | 617.9 | |
| GSC | 506.2 | 511.7 | 510.3 | |
| γCO | GSC | 640.9 | 658.9 | 655.2 |
| GAT | 563.3 | 575.5; 570.9 | 568.8 | |
| ASC | 490.3 | 501.7 | — | |
| δOCO | ASC | 636.5 | 622.8 | 624.7 |
| GSC | 628.6 | 646.1 | 644.8 | |
| GAT | 632.0 | 616.3 | 615.4 | |
| δC–CO | GAT | 491.4 | ≈496.9 | — |
| ASC | 477.8 | 490.0 | 477.5 (?) | |
| Inversion | GAT | 391.5 | 406.0 | — |
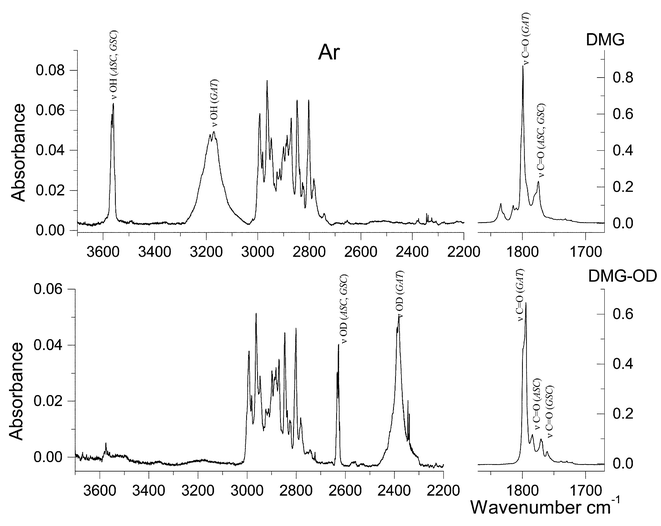 | ||
| Fig. 3 Infrared spectra of DMG and DMG-OD trapped in an argon matrix. Spectra obtained immediately after deposition at 9 K. Since the deuteration ratio was lower than 100%, “pure” DMG-OD spectrum was obtained by subtraction, so that OH stretching bands vanish. A narrow doublet at 2345/2339 cm−1 is due to matrix isolated CO2. | ||
 | ||
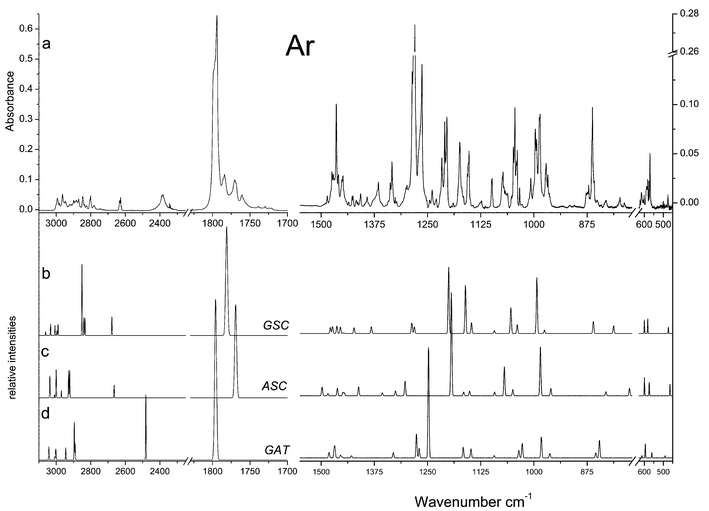
| Fig. 4 Relevant spectral regions of the infrared spectrum of DMG trapped in a xenon matrix (a: freshly prepared sample at 20 K; b: after annealing at 40 K), and calculated spectra for c: GSC, d: ASC and e: GAT conformers. The intensity scale of the calculated spectra in the 600–1180 cm−1 region is expanded twice when compared with that corresponding to the 1700–3850 cm−1 region. As mentioned in the Materials and methods section, annealing in Xe was performed up to a temperature of 60 K, but the spectroscopic changes observed in the temperature range 40–60 K follow the same trends as those observed in the range 20–40 K, illustrated in this figure, while the matrix optical properties start to deteriorate. | ||
 | ||
| Fig. 5 Relevant spectral regions of a: the infrared spectrum of DMG-OD trapped in an argon matrix (9 K) and calculated spectra for b: GSC, c: ASC and d: GAT conformers. The ratio between the intensity scales of the calculated spectra in the 1700–3100 cm−1 and 450–1550 cm−1 regions is 1.33. | ||
Fig. 3 displays the observed spectra of both DMG and DMG-OD in argon matrices. As expected, the spectra do not show any evidence of the presence of the zwitterionic species (e.g., no bands ascribable to the NH and carboxylate groups could be observed) and can be interpreted on the basis of the exclusive presence of neutral DMG molecules. The νO–H, νCO, δCOH, νC–O, τC–O, δOCO and γCO modes are characteristic vibrations and, according to the calculations, the corresponding bands should change appreciably in frequency upon deuteration. Hence, the bands due to these modes will be used as reference bands to help in the general assignment of the spectra. All the other bands lying in an accessible spectral region (>400 cm−1) are expected to be nearly coincident in DMG and DMG-OD spectra (see, for example, the CH stretching region of the spectra of the two compounds shown in Fig. 3: 3000–2700 cm−1).
Taking into consideration the theoretical results, in the experimental spectra of both isotopomers the most intense bands could be easily ascribed to the most stable GAT conformer, indicating that this form is the dominating species (see Tables 3 and S2 (ESI)† and Figs. 4 and 5). In addition, bands due to the ASC and GSC forms could also be identified in the spectra obtained in both argon and xenon matrices, as it will be shown in detail below.
The νOH stretching vibration is well known to be sensitive to hydrogen bonding.28,29 In proton donors, this mode usually gives rise to a broad feature in the 3400–3000 cm−1 spectral region, whilst in non-hydrogen-bonded matrix-isolated molecules this mode gives rise to a relatively sharp band at frequencies higher than 3500 cm−1. In the Ar matrices, the broad feature with maximum at ca. 3175 cm−1 can then be unequivocally assigned to νOH in the GAT conformer, showing the characteristic shape due to the involvement of the OH group in the OH⋯N bond. The complex feature observed at ca. 3566 cm−1 is here ascribed to the same mode in both the ASC and GSC forms (site splitted due to matrix site effects).
If we assume that the observed shift to lower frequency in the νOH stretching of the GAT conformer, relatively to the other two forms (ca. 391 cm−1, in the argon matrix), is only due to the involvement of its hydroxyl group in the OH⋯N bond, the enthalpy associated with this hydrogen bond may be estimated using the relationship derived by Rozenberg et al.,30
−ΔH=1.3(ΔνOH)1/2. The enthalpy obtained using this correlation and the experimental frequency data (in argon) is 25.7 kJ mol−1, a value that shows a fairly good agreement with that presented above, obtained using the −ΔHvs. N5⋯H4 calculated distance correlation. Note that the intramolecular hydrogen bond in DMG is considerably strong. For example, the observed shifts to lower frequencies in the νO–H mode due to intramolecular hydrogen bonding in glycolic (CH2OHCOOH), oxalic (HOOCCOOH) and pyruvic (CH3COCOOH) acids are much lower (80, 70 and 115 cm−1, respectively24,31,32). In malonic acid, whose most stable form was previously pointed out as possessing a quite strong intramolecular hydrogen bond, this shift is also smaller than in DMG (295 cm−125). In neutral glycine,9 the corresponding shift due to the intramolecular hydrogen bond is 360 cm−1, thus being of the same order of magnitude but still smaller than in DMG.
In DMG-OD, the νOD stretching bands are observed (splitted due to matrix site effects) at 2389/2382 cm−1 for the GAT conformer and in the 2627–2633 cm−1 region for ASC and GSC. These bands show the expected (theoretical) isotopic ratio (νOH/νOD ≈ √1.89 = 1.375), in consonance with the essentially localized nature of the νOH(D) mode. For both ASC and GSC the experimental isotopic ratio is 1.356, whereas the relatively stronger anharmonicity of νOH in the hydrogen bonded GAT conformer results in a decrease of the isotopic ratio for this vibration to 1.331. Note that the observed isotopic ratios closely follow those previously measured for matrix-isolated glycine, where the observed values were found to be 1.354 for the OH-free conformers and 1.338 for the OH⋯N hydrogen bonded conformer.9
In the carbonyl stretching region, the observed spectra (in argon) show an intense band with maximum at 1799 cm−1 (site splitted), which is due to the most stable conformer. The corresponding bands of the ASC and GSC forms are predicted to occur at lower frequencies and give rise to the features in the 1790–1760 cm−1 region. The precise nature of these bands could not be established unequivocally, and the proposed assignments (see Table 3) must be considered tentative (we shall return to this point later on). Minor bands due to traces of aggregates could also be observed in spectra of more concentrated matrices, in the 1760–1700 cm−1 region. The absence of these bands in the spectra shown in Fig. 3 confirms the origin of the above mentioned lower frequency carbonyl bands in the higher energy observed monomer conformations. In DMG-OD, the carbonyl stretching bands were observed at nearly similar frequencies as in the non-deuterated molecule, while, as expected, slightly deviated towards lower frequencies (see Figs. 3 and 5 and Tables 3 and S2 (ESI)†). In addition, in the spectrum of the deuterated compound, a better separation of the bands belonging to the highest energy conformers can be noticed. In consonance with the results of the calculations, the doublet appearing at ca. 1770 cm−1 is ascribed to the GSC conformer, while the bands appearing around 1760 cm−1 are assigned to conformer ASC. The small bands appearing above 1800 cm−1 in the spectra of the undeuterated molecule, which have no counterparts in DMG-OD, are ascribed to combination modes involving the τC–O(H) vibration as described in detail in Table 3.
The bands ascribable to the δCOH, νC–O, τC–O and γCO modes in the most abundant conformer are relatively easy to assign. In argon, δCOH gives rise to a site splitted Fermi doublet with component bands centred at 1388 and 1346 cm−1. The Fermi resonance is with all probability due to interaction with the τC–O+γCO combination band, whose fundamentals are observed at ca. 797 cm−1 and in the 570–575 cm−1 region, respectively. The νC–O stretching is predicted to be a considerably delocalized vibration (with important contributions from the C–C stretching and methyl rocking modes) of low intensity and gives rise to the band at 1190 cm−1. As expected, the τC–O torsion gives rise to a considerably broad and structured feature, which is observed in the 770–800 cm−1 region, with maximum intensity at 797 cm−1. The frequency of this mode may be correlated with that of the O–H stretching, since it is well known that a stronger hydrogen bond leads to a lower O–H stretching frequency and to a higher τC–O torsion.31–34 Indeed, the observed frequency for the hydrogen bonded conformer of DMG was found to fit well a linear plot of τC–O vs.νOH (Fig. 6) derived for a series of other carboxylic acids isolated in inert matrices, including several molecules that also exhibit intramolecular hydrogen bonds (formic, pyruvic, glycolic, oxalic, maleic, malonic25,31,32,34–36). This fact gives further support to the assignments made here, although this particular correlation is not a universal one, since besides hydrogen bonding other effects (e.g., different vibrational coupling and packing) might affect the frequencies, in particular that of the torsional mode. Finally, the carbonyl rocking mode in GAT is assigned to the feature near 575 cm−1 which appears as a doublet due to site splitting. In Xe, all the above discussed bands appear at nearly identical frequencies and are assigned straightforwardly—see Table 3.
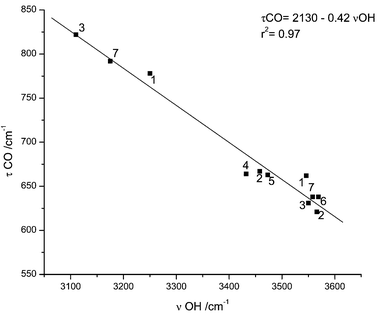 | ||
| Fig. 6 Plot of τCO vs.νOH frequencies for the series of carboxylic acids isolated in matrices: 1: malonic acid;25 2: oxalic;35 3: maleic;34 4: pyruvic;32 5: glycolic;31 6: formic;36 7: DMG (this work). | ||
The predicted shifts in the δCOH, νC–O, τC–O and γCO bands upon deuteration are −388, 95, −224 and −3 cm−1 respectively and, accordingly, the observed shifts (in argon) are ca.
−380, 81, −210 and −3 cm−1.
In the less abundant conformers of DMG, besides the carbonyl stretching mode, the νC–O, νC–N and δOCO modes are predicted to give rise to relatively intense bands lying in spectral regions where the dominant form does not absorb significantly. The predicted frequencies for these vibrations in the second most stable form (ASC) are 1114, 1064 and 636 cm−1, respectively, while in the third most stable conformer (GSC) the corresponding predicted values are 1105, 1052 and 629 cm−1. The observed spectra of DMG (both in argon and xenon matrices) clearly show bands in these spectral regions, further confirming the presence of more than one conformer in the matrices (see Figs. 3 and 4). However, since forms ASC and GSC are predicted to give rise to bands occurring at nearly the same frequencies, no precise assignments could be made only on the basis of the comparison between experimental and calculated data. Hence, in order to distinguish the spectroscopic features due to the two less abundant forms, annealing experiments were carried out both in argon and xenon matrices.
Annealing of the argon matrices is limited by the relatively small temperature range that can be used without significant loss of matrix properties (less than 30 K). The main spectroscopic changes observed in these experiments, upon increasing the temperature to 25 K, can be ascribed to conversion of less favourable matrix-sites to lower energy sites. In fact, noticeable changes in the relative intensities of the components of those bands that exhibit multiplet structure due to trapping of a given conformer in a different local environment (matrix site splitting) were observed. The non-observance of any conformer interconversion process is consistent with the estimated high energy barriers for these reactions (note that no conformational interconversions were also observed upon the attempted in situ broad band infrared irradiation of the matrices). Aggregation was also observed, with appearance of aggregates characteristic bands, e.g. in the low frequency range of the carbonyl stretching region and decrease of bands due to monomers.
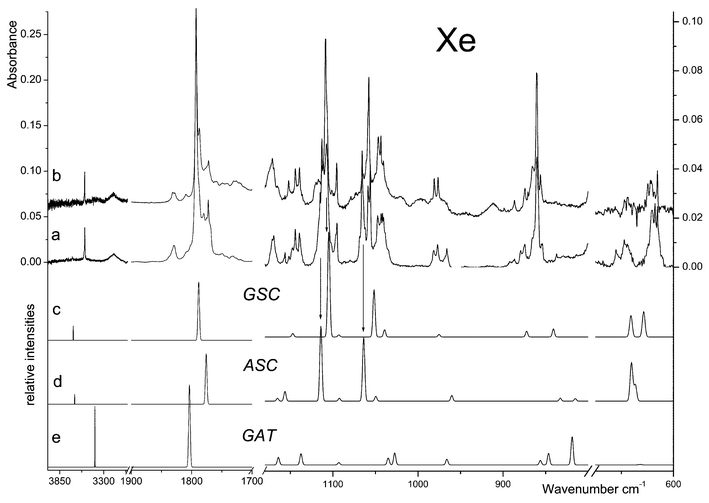
Annealing of the xenon matrices yielded much more interesting results, allowing us to attain a more clear picture of the system under study, as described in detail below. As for the argon experiments, two different phenomena could be observed: (i) site conversion and (ii) aggregation. Again, no conformational isomerizations were observed, as it could be anticipated taking into consideration the predicted energy barriers for interconversion between the various conformers (see Table 2). The spectral changes accompanying aggregation in the Xe matrix were found to be particularly useful to shed light on the spectral signature of the ASC and GSC forms of monomeric DMG.
In Fig. 4, the spectrum of DMG in a xenon matrix, prepared from deposition of DMG vapours at T≈323 K onto the cold substrate of the cryostat kept at 20 K, is compared with the spectrum after annealing of the matrix up to 40 K. The most relevant differences were observed in the 1200–600 cm−1 region and accompany the appearance of the typical bands of aggregates in the 1760–1720 cm−1 region. All the observed changes were found to be irreversible (a subsequent re-cooling of the matrix did not produce any observable changes). At all temperatures covered during the annealing (20–60 K) no evidence was found supporting the presence of zwitterionic species in the matrix. In particular, no bands were observed in the characteristic νNH stretching region. Taking also into consideration the results of the calculations, it can be concluded that those bands that better fit the calculated spectrum of the ASC conformer suffer a pronounced decrease in intensity in favour of the aggregates, while the bands ascribable to GAT and GSC appear to be considerably less affected by aggregation. These results seem to indicate that aggregation of DMG in the xenon matrix occurs preferentially for DMG in the ASC conformation. It is worthwhile mentioning that, at the highest temperature reached in these experiments the bands due to the ASC conformer almost disappeared from the spectrum. This is clearly observed, for example, in the case of the bands at ca. 1113 cm−1
(νC–O; site splitted) and 1066 cm−1
(νC–N; also site splitted), which are due exclusively to this conformer. In the case of the carbonyl stretching, however, this behaviour could not be so clearly noticed due to the overlap of the bands originated in the ASC and GSC forms as well as to the fact that additional bands due to aggregated species start to contribute significantly to the observed spectral profile in this region (see Fig. 4). In any case, the subtraction of the spectrum obtained for the freshly prepared Xe matrix from that of the annealed (37 K) sample reveals that, in this region, the features that reduce more in intensity upon annealing appear at 1780, 1772 and 1770 cm−1, being here ascribed to the ASC form (the 1801 cm−1 band follows an identical behaviour and was attributed to the τC–O+νN–C combination tone originated in ASC—see Table 3).
It is not surprising that the GAT conformer, in which both OH and amino groups are involved in a strong intramolecular hydrogen bond, is less able to aggregate than the ASC and GSC forms. On the other hand, a priori, the relative ability of the two higher energy conformers to aggregate could not be expected to be markedly different, since both forms have the relevant groups available for participation in intermolecular hydrogen bonding interactions. However, there are several structural differences between these forms that might explain the observed different behaviour regarding their relative ability to aggregate. Firstly, the ASC conformer has both a slightly larger positive charge on the hydroxyl hydrogen and a larger negative charge on the carbonyl oxygen than GSC
(0.277 vs. 0.283 and −0.293 vs.
−0.301 e, respectively) (MP2 (6-31++G**) and DFT/B3LYP (6-311++G**) calculated Mulliken charges on atoms for the three most stable conformers of DMG are shown in Table S10 (ESI)†; 1e=1.6×10−19 C), i.e., the OH group is more acid and the carbonyl group is a better proton acceptor in the conformer that shows a greater trend to aggregate. Moreover, ASC has a higher symmetry than GSC exhibiting a less skewed structure and it has an electron spatial extent 〈R2〉1/2 that is relatively smaller than those of the other two conformers (1534 pm vs. 1591 pm in GSC and 1562 pm in GAT). Hence it can be expected to perturb to a lesser extent the packing in the matrix, in particular when we consider dimeric or higher order polymeric structures. In addition, the nitrogen lone electron pair is in a more accessible position to participate in intermolecular interactions in ASC than in GSC. This may be important when larger associates are considered.
The energies of the possible carboxylic cyclic dimers based on either ASC or GSC monomers were calculated at the DFT(B3LYP)/6-311++G**. In consonance with the matrix-isolation annealing experiments, the dimer based on ASC was found to be more stable than those based on GSC by more than 4 kJ mol−1. The ASC-based dimer that was considered here has a centrosymmetric geometry; in the case of GSC, two dimers were studied: (i) the centrosymmetric dimer, where the gauche methyl groups linked to the N atom in the two constituting monomers are in opposite sides of the hydrogen bond ring plane (ΔE=4.36 kJ mol−1), and (ii) the alternative dimeric structure where the gauche methyl groups are located in the same side of the ring plane (ΔE=4.39 kJ mol−1). In addition, as discussed above, it can be expected that the ASC-based dimer gain an additional stabilization in the matrices relative to those constituted by GSC units due to better packing. Besides, the calculated electron spatial extent 〈R2〉1/2 for the ASC-based dimer is only 3857 pm, while the same quantity in both GSC dimers are ca. 4070 pm. All these data are consistent with a faster aggregation by the ASC molecules, as observed experimentally.
The energy of the hydrogen bonds in the dimers can also be estimated using Rozenberg's empirical correlation.30 Taking into consideration the calculated hydrogen bond distances, an average value of ca.
−30 kJ mol−1 is obtained, which is within the usual range of energies of this interaction in carboxylic acid dimers.37,38 Note that the value obtained using the Rozenberg's correlation agrees very well with the H-bond energies derived from the DFT calculated dimerization energies. For ASC, the dimerization energy (Edimer−2×EASC) amounts to ca.
−64 kJ mol−1, while for the two dimers of GSC the corresponding energies were predicted to be ca.
−60 kJ mol−1. Since two hydrogen bonds exist in the dimers, the H-bond energies can be easily estimated from dimerization energies, being ca.
−32 and −30 kJ mol−1, respectively for ASC and GSC, in good agreement with the empirically determined value presented above.
The fact that, contrary to what was found in xenon, in the argon matrices the relative abilities of the different DMG conformers to aggregate do not appear to be considerably different, must be a consequence of changes (upon varying the matrix gas) in the interactions between the solute molecules and the matrix that are relevant to the aggregation process. The different size of argon and xenon atoms (leading to a larger number of argon atoms in the primary solvation sphere and to different packing) and the different rigidity and polarizability of the two matrices do certainly play a role in determining the different observed behaviour.
Once the nature of the three conformers that contribute to the spectra of DMG was established, their relative energies could be estimated from the band intensities, weighted by the corresponding calculated intensities. In this estimation, we assume that populations corresponding to gas phase equilibrium at a temperature of ≈323 K (the temperature of the mini oven in our set up) were efficiently trapped. Bands in the OH stretching region and at ca. 575 and 967 cm−1
(in argon) were used since they lie in relatively clean spectral regions. The values estimated by this way (ΔEASC–GAT=2 kJ mol−1, ΔEGSC–GAT=4 kJ mol−1) compare fairly well with the DFT calculated values for the molecule in vacuum (ΔEASC–GAT=2.7 kJ mol−1, ΔEGSC–GAT=5.4 kJ mol−1—see also Table 1).
Conclusion
Matrix-isolated infrared spectroscopy supported by theoretical predictions undertaken at the DFT(B3LYP)/6-311++G** and MP2/6-31++G** levels of theory allowed, for the first time, unequivocal observation and spectral signature characterization of three conformers of DMG. Assignment of the observed spectra (both in Ar and Xe) was carried out on the basis of comparison with the theoretical spectra, annealing experiments and isotopic substitution.The conformational ground state was found to be the intramolecularly O–H⋯N hydrogen-bonded GAT form, where the Lp-N–C–C and N–C–CO dihedral angles are 30° and ca. 180°, respectively, and the carboxylic group assumes the trans configuration. In the second (ASC) and third (GSC) lower energy forms of DMG, the carboxylic moiety assumes the cis conformation while the N–C–CO axis adopts the syn arrangement and the conformation around the N–C bond is respectively anti and gauche.
The energy barriers for conformational interconversion between the three experimentally observed conformers were found to be considerably large, thus preventing the occurrence of these processes in the matrices.
Aggregation in the xenon matrix-isolated species was found to be dependent on the conformational state of the monomers, the ASC conformer exhibiting a considerably higher ability to aggregate than both the GAT and GSC forms.
Acknowledgements
This work was supported by the portuguese Fundação para a Ciência e a Tecnologia (Research Project POCTI/QUI/43366/2001 and Grant FCT #SFRH/BPD/1661/2000). A.G.-Z. acknowledges the post-doctoral grant from the Instituto para a Cooperação Científica e Tecnológica Internacional (ICCTI), Lisbon.References
- R. Binzak, S. Wevers, Y. Moolenaar, W. Lee, J. Hwu, U. Poggi-Bach, H. Engelke, J. Hoard, Joseph Vockley and Jerry Vockley, Am. J. Hum. Genet., 2001, 68, 839 CrossRef.
- M. Laryea, F. Steinhagen, S. Pawliczek and U. Wendel, Clin. Chem., 1998, 44, 1937 Search PubMed.
- V. Rodrigues, J. Paixão, M. Costa and A. Matos Beja, Acta Crystallogr., Sect. C, 2001, 57, 417 CrossRef CAS.
- K. Hariganesh and J. Prathiba, J. Pharm. Pharmacol., 2000, 52, 1519 Search PubMed.
- R. C. Weiss, Am. J. Vet. Res., 1992, 53, 829 Search PubMed.
- N. von Weymarn, A. Nyyssölä, T. Reinikainen, M. Leisola and H. Ojamo, Appl. Microbiol. Biotechnol., 2001, 55, 214 Search PubMed.
- M. L. Mendum and L. T. Smith, Appl. Environ. Microbiol., 2002, 68, 813 CrossRef CAS.
- T. van Der Heide and B. Poolman, J. Bacteriol., 2000, 182, 203 CAS.
- S. G. Stepanian, I. D. Reva, E. D. Radchenko, M. T. S. Rosado, M. L. T. S. Duarte, R. Fausto and L. Adamowicz, J. Phys. Chem., 1998, 102, 1041 CrossRef CAS.
- S. G. Stepanian, I. D. Reva, E. D. Radchenko and L. Adamowicz, J. Phys. Chem. A, 2001, 105, 10664 CrossRef CAS.
- A. Headley and S. Starnes, J. Mol. Struct. (THEOCHEM), 1996, 370, 147 CrossRef CAS.
- A. Headley and S. Starnes, J. Mol. Struct. (THEOCHEM), 1998, 453, 247 CrossRef CAS.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, V. Zakrzewski, J. Montgomery, R. Stratmann, K. Burant, S. Dapprich, J. Millam, A. Daniels, K. Kudin, M. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. Petersson, P. Ayala, Q. Cui, K. Morokuma, D. Malick, A. Rabuck, K. Raghavachari, J. Foresman, J. Cioslowski, J. Ortiz, A. Baboul, B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. Martin, D. Fox, T. Keith, M. Al-Laham, C. Peng, A. Nanayakkara, M. Challacombe, P. Gill, B. Johnson, W. Chen, M. Wong, J. Andres, C. Gonzalez, M. Head-Gordon, S. Replogle and J. Pople, Gaussian 98, revision A.9, Gaussian Inc., Pittsburgh, PA, 1998.
- M. J. Frisch, M. Head-Gordon and J. A. Pople, Chem. Phys. Lett., 1990, 166, 281 CrossRef CAS.
- P. Csaszar and P. Pulay, J. Mol. Struct. (THEOCHEM), 1984, 114, 31 CAS.
- J. H. Schachtschneider, Technical Report, Shell Development Co., Emeryville, CA, 1969.
- C. Peng and H. B. Schlegel, Isr. J. Chem., 1994, 33, 449 Search PubMed.
- A. V. Iogansen and M. Rozenberg, J. Appl. Spectrosc. (USSR), 1968, 9, 1027 Search PubMed.
- J. J. C. Teixeira-Dias and R. Fausto, J. Mol. Struct., 1986, 144, 199 CrossRef CAS.
- R. Fausto, F. P. S. C. Gil and J. J. C. Teixeira-Dias, J. Chem. Soc., Faraday. Trans. 2, 1993, 89, 3235 Search PubMed.
- R. Fausto, J. Mol. Struct. (THEOCHEM), 1994, 1315, 123 CrossRef.
- R. Fausto, A. G. Martins, J. J. C. Teixeira-Dias, P J. Tonge and P. R. Carey, J. Phys. Chem., 1994, 98, 3592 CrossRef CAS.
- S. Stepanian, I. D. Reva, E. D. Radchenko, M. T. S. Rosado, M. L. T. S. Duarte, R. Fausto and L. Adamowicz, J. Phys. Chem., 1998, 102, 1041 CrossRef CAS.
- R. Fausto and E. M. S. Maçôas, J. Mol. Struct., 2001, 563/564, 29 CrossRef.
- E. M. S. Maçôas, R. Fausto, J. Lundell, M. Pettersson, L. Kriachtchev and M. Rasanen, J. Phys. Chem. A, 2000, 104, 11725 CAS.
- R. Fausto, P. R. Carey and P. J. Tonge, J. Chem. Soc., Faraday Trans., 1994, 90, 3491 RSC.
- V. Alexandrov, S. Stepanian and L. Adamowicz, Chem. Phys. Lett., 1998, 291, 110 CrossRef CAS.
- C. Cacela, M. L. Duarte and R. Fausto, Vib. Spectrosc., 2001, 26, 113 CrossRef CAS.
- C. Cacela, M. L. Duarte and R. Fausto, Spectrochim. Acta, Part A, 2000, 56, 1051 Search PubMed.
- M. Rozenberg, A. Loewenschuss and Y. Marcus, Phys. Chem. Chem. Phys., 2000, 2, 2699 RSC.
- S. Jarmelo, T. M. R. Maria, M. L. P. Leitao and R. Fausto, Phys. Chem. Chem. Phys., 2001, 3, 387 RSC.
- I. Reva, S. Stepanian, L. Adamowicz and R. Fausto, J. Phys. Chem., 2001, 105, 4773 CrossRef CAS.
- M. Rozenberg, Spectrochim. Acta Part A, 1996, 52, 1559 CrossRef.
- E. M. S. Maçôas, R. Fausto, J. Lundell, M. Pettersson, L. Kriachtchev and M. Rasanen, J. Phys. Chem. A, 2001, 105, 3922 CrossRef CAS.
- E. M. S. Maçôas, R. Fausto, J. Lundell, M. Pettersson, L. Kriachtchev and M. Rasanen, J. Phys. Chem. A, 2000, 104, 6956 CrossRef CAS.
- R. Fausto, L. A. E. Batista de Carvalho, J. J. C. Teixeira-Dias and M. N. Ramos, J. Chem. Soc., Faraday Trans. 2, 1989, 85, 1945 RSC.
- A. V. Iogansen, Spectrochim. Acta, Part A, 1999, 55, 1585 Search PubMed.
- S. Lifson, A. T. Hagler and P. Dauber, J. Am. Chem. Soc., 1979, 101, 5111 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables (Tables S1–S10) and figures (Figs. S1 and S2) of supporting information. See http://www.rsc.org/suppdata/cp/b2/b207320j/ |
| This journal is © the Owner Societies 2003 |