On the origin of binding energy shifts of core levels of supported gold nanoparticles and dependence of pretreatment and material synthesis
Jörg
Radnik
*a,
Christian
Mohr
b and
Peter
Claus
b
aInstitute for Applied Chemistry, Berlin-Adlershof e.V., Richard-Willstätter-Str. 12, D-12489 Berlin, Germany. E-mail: radnik@aca-berlin.de
bDepartment of Chemistry, Institute of Chemical Technology II, Darmstadt University of Technology, Petersenstr. 20, D-64287 Darmstadt, Germany
First published on 14th November 2002
Abstract
X-ray photoelectron spectroscopy (XPS) investigations of supported nanoparticles smaller than 10 nm show a significant shift of the electron binding energy of core levels compared with the bulk values. In this work, such shifts were examined at differently supported and prepared gold nanoparticles for the 4f electron level. Special attention was paid to the influence of reducing pretreatment in hydrogen and, moreover, the influence of different oxide supports. Surprisingly, in most cases, lower binding energies than the Au 4f7/2of 84.0 eV were observed depending on the oxidic support as well as the pretreatment conditions. The origin of these differences of the core level values are discussed in terms of different models like electron transfer from the support to the particles, size and geometric effects. It seems that especially geometric factors like the particle shape play an important role.
1. Introduction
In the last years, supported gold catalysts with particle sizes smaller than 20 nm have attracted a lot of attention. While gold bulk metal is rather inert in chemical reactions and thus is of low interest for catalysis, highly dispersed gold catalysts show surprising high activities in some reactions important for chemical industries. The oxidation of carbon monoxide at low temperatures is mainly used as model reaction to investigate the catalytic potential of gold.1 Furthermore, we developed supported gold catalysts for the hydrogenation of acrolein as α,β-unsaturated aldehyde to allyl alcohol.2Especially concerning CO oxidation, numereous investigations were carried out. However, the nature of the active site is still unclear and controversially discussed. Using scanning tunneling microscopy/spectroscopy study combined with reaction kinetics measurements, Valden et al., studying model catalysts, related the catalytic properties to a quantum size effect with respect to the thickness of gold clusters.3 Other investigations give hints for charged or oxidized gold particles as active sites, whereas both negatively and positively charged particles were found.4–7
Other authors argue against an electronic effect in small Au particles as the major factor contributing to the activity of supported gold catalysts. The Au–support interface8,9 or a phase of the support modified by the gold nanoparticles was supposed to be the location of the active sites.10
A suitable method to investigate electronic properties of nanoparticles is X-ray photoelectron spectroscopy (XPS). From the electron binding energy the oxidation state of a component can be obtained.11 It is principally necessary to distinguish between initial state effects depending mainly on the orbital energy of the emitted electron, and final state effects reflecting the relaxation energy of the system, which corresponds to the energy difference between the excited atom after loss of an electron and the relaxed electron system, which results from a minimization of its total energy.
For nanoparticles, core level shifts due to the particle size must be considered in the interpretation of such spectra.12 Initial effects like the intrinsic size effect caused by the reduced average coordination number of surface atoms in small particles lead to decreasing binding energy for the core level electrons. Furthermore, electron exchange with the support can change the binding energy of the electrons emitted from the nanoparticle. The binding energy is also influenced by the relaxation energy. These “final-state” effects depends on the particle size, too.12
Several XPS investigations have been carried out focussing mainly on gold as oxidation catalyst. In a comparative study of Au/TiO2 and Au/ZrO2 used as catalysts for CO oxidation by Grunwaldt et al., XPS investigations indicated a metallic state of the gold. CO adsorption experiments show significant sensitivity to structure, which was attributed to different rounding of the gold particles, depending on support and pretreatment, which affected the number of low-coordinated gold atoms.13 Possible explanations of these different results might be differences in the preparation and pretreatment of the catalysts.
In the work presented here, the influence of the kind of oxide support and of the specifics of preparation on the electronic properties of the Au nanoparticles has been investigated. To get information about particle–support interactions we focussed our study on samples with low Au loading (<0.2 at.% in the near-surface region). In the first part of this paper, results of samples pretreated differently are presented. In the second part, the influence of both the method of sample preparation as well as the kind of support are examined. Special attention was paid to the influence of pretreatment in hydrogen, which has never been the subject of XPS studies on gold catalysts before. For a better understanding of the dependence of the XP spectra on the structure of the gold particles, these spectra are related to transmission electron microscopy (TEM) results. For a further investigation of structural effects on the spectra, indium was added to the gold particles, which makes it possible to distinguish the electron binding energy of atoms at low coordinated sites from those in the terrace.
2. Experimental
The details of sample preparation by impregnation (I), coprecipitation (P) and deposition–precipitation (DP) are given elsewhere.14,15 Additionally, samples prepared by chemical vapor deposition (CVD) as described by Schimpf et al.16 were used. The metal content was about 2 wt.%. The structural characterization of the reduced catalysts, especially the investigation of shape and morphology of gold particles as well as the crystal structure of the supports, was realized by application of transmission electron microscopy (TEM). While for conventional TEM a combination of bright field, dark field and selected area electron diffraction on a JEOL JEM 100 C (100 kV) was used, for high-resolution TEM (HRTEM) a JEM 4000 (400 kV) was taken.The XP spectra were obtained with a VG ESCALAB 220 iXL equipped with a monochromated Al Kα radiation source. For charge compensation a flood gun with variable electron voltage (from 6 to 8 eV) was used and a variable emission current was applied. The emission current was varied from 0.10 to 0.25 mA, depending on the sample. The spectra were referenced to the main peak of the cationic compound of the support (Ti 2p3/2 from TiO2: 458.8 eV, Zr 3d5/2 from ZrO2:182.8 eV, Si 2p from SiO2: 103.3 eV, Zn 2p3/2 from ZnO: 1022.3 eV). The peak positions could be determined with a precision of ±0.1 eV. On the samples Au/TiO2-CVD and Au/SiO2-CVD examined in the first part, several pretreatment procedures (i.e. heating in flowing air as well as flowing hydrogen in a temperature range between 573 K and 723 K) were carried out. In the second part, the electronic properties of gold particles on different oxide supports are compared after treatment in a hydrogen flux at 573 K. The treatment both in hydrogen as well as in air before the XPS measurements were realized in a gas cell integrated in the lock of the system at the desired temperature and in the gas flux. Thus, it was possible to transfer the sample from this gas cell to the UHV analysis chamber without air contact. The pressure in the analysis chamber was below 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ×10−6 Pa.
×10−6 Pa.
3. Results
3.1 Influence of the pretreatment conditions
First, investigations of the influence of the preparation conditions on the XP spectra of the gold samples were carried out. The spectra of the sample Au/TiO2-CVD are shown in Fig. 1. On the freshly prepared sample, the Au 4f7/2 binding energy has a value of 83.6 eV (Fig. 1, spectrum (a)). After pretreatment in a hydrogen flux at 573 K a shift to energies lower by 0.3 eV was observed (spectrum (b)). This was followed by a treatment in air at 673 K for 2 h, resulting in a binding energy of Au 4f7/2 at 83.8 eV (spectrum (c)). Subsequent oxidation in air at 773 K had no influence on the XP spectra (not shown). A subsequent treatment of the sample with hydrogen at this temperature led to a small shift of the Au 4f7/2 energy to smaller values which is in the error range of the binding energy (83.7 eV, (spectrum (d)). As a result of a subsequent oxidation in air at 773 K, a Au 4f7/2 peak at 84.4 eV, together with an additional small broad one around 86 eV was found (spectrum (e)).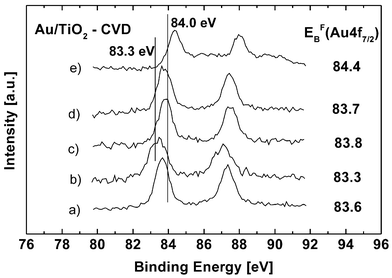 | ||
| Fig. 1 Influence of the pretreatment on the Au 4f7/2 binding energy of a Au/TiO2 catalyst after different pretreatments: (a) as synthesized with chemical vapor deposition, (b) after subsequent reduction in a hydrogen flux at 573 K for 2 h, (c) after 2 h in an air flux at 673 K, (d) and additional 2 h in an air flux at 773 K and 2 h in a hydrogen flux at 773 K, (e) and 2 h in an air flux at 773 K. The maxima of the Au 4f7/2 peaks are listed. The error range is ±0.1 eV. | ||
The latter spectrum can be explained by gold partially oxidized to Au2O3. Generally, hydrogen pretreatment lowers the binding energy, whereas an increase of binding energies was observed after treatment in air.
A representative TEM image of the sample Au/TiO2-CVD after treatment in hydrogen at 673K is shown in Fig. 3. From TEM measurements, the mean gold particle size (2.7 nm) and the mean square displacement (1.4 nm) were derived.
For Au/SiO2-CVD, the same series of pretreatment experiments were carried out as for Au-TiO2-CVD. The results are summarized in Fig. 2. Without pretreatment, similar values for gold supported on TiO2 and SiO2, respectively, were observed. This remains true for the evolution of the Au 4f7/2 binding energy after the different pretreatments with hydrogen and air, whereas on SiO2, the negative shifts are more significant than on TiO2. However, the halfwidth of the Au 4f peaks for this system is significantly higher. It is still unclear, whether this is an effect of the relaxation time of the excited state with one electron fewer or of greater heterogeneity in the chemical environment of the nanoparticles.
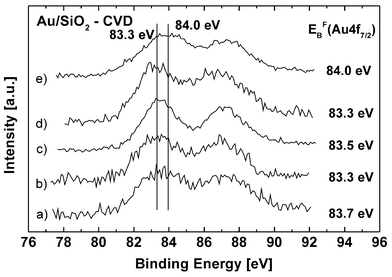 | ||
| Fig. 2 Influence of the pretreatment on the Au 4f7/2 binding energy of a Au/SiO2 catalyst after different pretreatments: (a) as synthesized with chemical vapor deposition (binding energy 83.7 eV), (b) after subsequent reduction in a hydrogen flux at 573 K for 2 h (83.2 eV), (c) after 2 h in an air flux at 673 K (83.3 eV), (d) and additional 2 h in an air flux at 773 K and 2 h in a hydrogen flux at 773 K (83.0 eV), (e) and 2 h in an air flux at 773 K (83.8 eV). The maxima of the Au 4f7/2 peaks are listed. The error range is ±0.1 eV. | ||
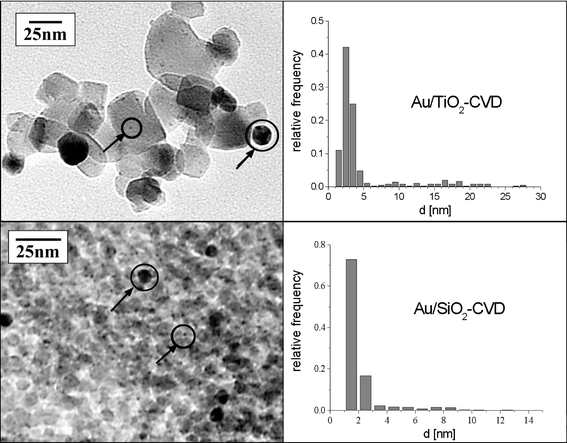 | ||
| Fig. 3 TEM overview (left) and gold particle size distribution of the catalyst Au/TiO2-CVD (upper row) and Au/SiO2-CVD (lower row). In the TEM images gold particles are indicated by arrows. | ||
3.2 The evolution of Au binding energy after hydrogen pretreatment on different TiO2 supported samples
Prior to the XPS studies, all samples were treated in situ in a flux of hydrogen within the gas cell. A typical XP spectrum for highly dispersed Au particles on TiO2 is shown in Fig. 4, which is derived from Au/TiO2-SG. The mean size of the gold particles estimated by TEM was 1.1 nm.14 After preparation in the gas cell at 723 K, an electron binding energy of the Au 4f7/2 transition at 83.4 eV was obtained (Fig. 4). The spectra of gold titania catalyst prepared by other methods (i.e. DP and I) look similar. The estimated values of binding energies are summarized in Table 1. The small changes of binding energy values for different samples based on the same support (TiO2) indicate negligible influence of the preparation method on the electronic properties of the gold particles. To verify this negative core level shift observed for Au nanoparticles with diameters markedly below 10 nm, these spectra were compared with those of a sample established as a reference in XPS data analysis.17 The latter had been prepared by impregnation of TiO2 with a colloidal solution of well-defined gold particles having a mean particle diameter of around 20 nm. After correcting the Ti2p3/2 value to 458.8 eV (the value expected for titania), a Au 4f7/2 binding energy of 84.0 eV was observed, which is the value for metallic gold.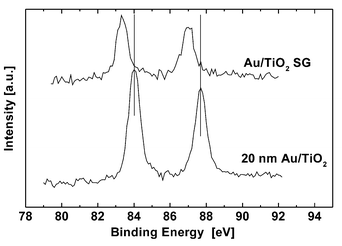 | ||
| Fig. 4 Comparison of the Au 4f7/2 spectra of Au/TiO2-SG and a XPS reference sample, consisting of gold particles (mean diameter 20 nm) on TiO2. The values for bulk gold are marked with lines. | ||
| Sample | Binding energy (Au 4f7/2)/eV | d Au/nm | msd/nm | Reference of TEM analysis |
|---|---|---|---|---|
| Au/TiO2–I | 83.0 | 2.0 | 0.4 | 15 |
| Au/TiO2–DP | 83.3 | 5.3 | 0.3 | 15 |
| Au/TiO2–SG | 83.4 | 1.1 | 0.2 | 15 |
| Au/TiO2–CVD | 83.3 | 2.7 | 1.4 | This work |
| Au/SiO2–CVD | 83.2 | 1.4 | 0.3 | This work |
| Au/SiO2–I | 83.2 | 3.8 | 2.3 | 14 |
| Au/ZrO2–F | 83.8 | 1.4 | 0.3 | 15 |
| Au/ZnO–I | 84.4 | 9.0 | 0.3 | 18 |
| Au-In/ZnO–I | 83.8 | 10.1 | 0.2 | 18 |
3.3 Influence of the hydrogen pretreatment on the support
To study the effect of hydrogen treatment on the support itself, high resolution XP spectra of the Ti 2p peaks were measured. After hydrogen pretreatment, the Ti 2p peaks of the gold/titania system show only one oxidation state, namely Ti(IV). In contrast, the Ti 2p3/2 peak of pure titania exhibits a small shoulder at lower binding energies which can be related to Ti(III) (Fig. 5). This can be explained by a small reduction of the Ti atoms of pure titania from +IV to +III under the reducing pretreatment conditions. | ||
| Fig. 5 Ti 2p spectra of gold-deposited and pure titania after hydrogen pretreatment at 723 K. The surrounding area is magnified. The position of the Ti 2p3/2 peak of Ti(III) is marked by a line. | ||
3.4 Influence of the kind of support
To get insights into the nature of the interactions between gold particles and supports, the latter were varied. TiO2, SiO2, ZrO2 and ZnO were compared here and the results of the different measurements after hydrogen pretreatment were statistically evaluated (Table 1). No differences in the spectra were found between the catalysts treated with hydrogen at 573 K and 723 K. However, a clear dependence of the Au 4f7/2 binding energy on the kind of oxide support was observed. The lowest Au 4f7/2 electron binding energy levels were found for SiO2 and TiO2 supported catalysts. In contrast, for gold particles deposited on ZrO2 a binding energy of 83.8 eV (close to 84.0 eV, the value of metallic bulk gold) was observed. For the system Au/ZnO-I, the binding energy is slightly higher than for metallic gold (84.4 eV).3.5 Adding of indium to the gold nanoparticles
Recently, we investigated the influence of adding indium to a Au/ZnO-I catalyst after reduction in flowing hydrogen at 573 K by means of HRTEM.18 We were able to show that this resulted in a selective deposition of indium on the faces of the gold particles (as well as on the interface between gold and support), leaving just the edges of the cuboctahedral gold particles unmodified.Moreover, the existence of mixed phases containing both gold as well as indium could be excluded. Nevertheless, the addition of indium to Au/ZnO-I has also an influence on the XP spectra of gold, i.e. this modification was accompanied by a pronounced shift of the Au 4f7/2 electron binding energy: For the monometallic system Au/ZnO-I, a value of 84.4 eV was measured, while for the bimetallic Au–In/ZnO-I catalyst the binding energy decreased to a value of 83.8 eV (Fig. 6).
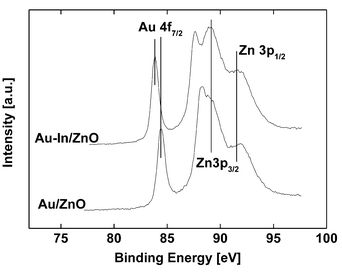 | ||
| Fig. 6 Au 4f spectra of Au/ZnO and Au–In/ZnO after hydrogen pretreatment at 573 K. Next to the Au 4f peak system the Zn 3p peaks are presented. | ||
4. Discussion and model
The experiments clearly show the strong influence of pretreatment conditions on the binding energy of Au electrons ejected from small particles. Additionally, the kind of oxide support used plays an important role. For nanosized gold particles on SiO2, TiO2 and ZrO2, pretreatment in hydrogen leads to a shift of the binding energy to lower values.Before discussing the physical and chemical origin of these shifts, artefacts like charging effects must be excluded. A problem with such samples (i.e. small particles on an insulating support) may be differential charging. This is excluded here because the half width of the peak is very small (ca. 0.75 eV) and shows no broadening, which is a hint for the absence of charging effects. Moreover, because of the higher total photoionisation cross section of the metal atoms in comparison to the support atoms, positively charged metal atoms with higher electron binding energies compared to the bulk value could occur. The same was observed for small particles on insulators due to the incomplete screening of the electrons holes in the final state.12 Since the binding energy values of gold decrease in all samples, the occurrence of these effects is excluded.
Sometimes, negative binding energy shifts were observed for large gold particles (diameter above 5 nm) having bulk properties and an separate Fermi edge due to the decoupling of the Fermi edge of the metal particle from that of the spectrometer.19 Under the measurement conditions applied here, this is not the case, which is demonstrated by the measurements on the reference sample having a gold particle size of 20 nm. Thus, the decrease of binding energy of the Au electrons in nanosized gold particles is not artefactual and should have a physical origin.
Such negative shifts may be related to different effects, namely an influence of the oxidation (electronic) state of the Au particles, particle size effects or different structural arrangement in the surface of the particles. These different effects are discussed in the following subsections.
4.1 Oxidation (electronic) state of gold
A possible explanation of the low binding energy of the Au particles after hydrogen pretreatment could be electron transfer from the support to the gold nanoparticles, accompanied by negative charging of the particles. Investigations of the TiO2 support itself may suggest such an assumption. For pure TiO2, a small quantity of reduced Ti species, probably Ti3+, was found after the pretreatment in hydrogen at 723 K (see Fig. 5). For Au/TiO2, no evidence for such reduced Ti species were observed. The low value of the electron binding energy of gold could indicate a negative charge. In fact, SMSI effects (strong support–metal interactions) are well-known phenomena occurring in systems consisting of metal particles deposited on reducible supports like TiO2. In such cases, e.g. for Pt/TiO2, negative shifts of the binding energy of were measured via XPS after a high-temperature reduction in hydrogen at 773 K.20 However, the shift to lower BE values occurred already at 573 K, which is distinctly below the temperature of 700 K, which is generally required for the occurrence of SMSI effects.In recent publications, Sanchez et al.21 as well as Claus et al.15 proposed f-centres as electron donating sites in the oxide support. These f-centres are oxygen vacancies within the support filled with electrons and their concentration varies with the kind of support. In ab initio simulations, Sanchez et al. showed for MgO a charge transfer from the oxide support to the Au particles for both, a defect-free and a surface containing f-centres. However, the binding energy for a gold particle near a defect site was significantly higher than on the defect-free surface. The authors remark that the charging of the adsorbed gold particles plays an essential role in the enhanced binding to the surface and localize them near defect sites. In a recent ESR study, signals of reduced species Ti3+ or Zr3+ were observed for Au on TiO2 and ZrO2 samples.15 Furthermore, the existence of f-centres in some of these gold catalysts was shown. The amount of both reduced species and f-centres increased after hydrogen treatment. These supports were also used in this study.
A dependence of electronic interaction between particle and support on the amount of reduced species (and/or f-centres) could be possible, since on supports with lower reducibility, as ZrO2 and ZnO, the charge of the gold particles is expected to be not strongly influenced by the support. However, the low binding energy of Au4f7/2 electrons on Au/SiO2 is not understandable in this context, since SiO2 is known for its weak interaction with noble metal clusters.22
For the TiO2 support, the following redox reaction may be proposed: Ti3++Au→Ti4++Au−. However, such a redox process has been excluded by Zwijnenburg et al.23 In their recent XPS investigations, the binding energy of the Au 4f7/2 electrons of Au nanoparticles supported on TiO2 was below 84.0 eV, i.e. below the value of metallic gold, which is in agreement with our experimental results. However, only metallic gold and no hints of negatively charged Au atoms were found by XPS as well as Mössbauer spectroscopy. Thereby, the existence of metallic gold via XPS was verified via analysis of the Auger parameter. The latter was not possible for the samples examined here, due to the much lower metal content (generally about 2 wt.%) compared to the sample used by Zwijnenburg et al.
(metal content about 10 wt.%).
4.2 Particle size effects
For Au/TiO2, the difference of particle sizes (5.5 nm and 1.1 nm, respectively) for different samples shows no significant influence on the binding energy of the Au4f7/2 electrons emitted from these particles, even though the size dependence of initial and final state effects is well known, hence the shift of core levels and higher binding energy values generally. Due to the small content of gold and the small cross section for the Au MNN or NOO Auger transition, it was not possible to measure the Auger parameters and to distinguish experimentally between the influence of initial and final state effects. Because of these experimental problems, the binding energy shift originated by these effects could be estimated only roughly. For an isolated spherical cluster, the final state effect is analyzed simply by assuming that a single charge is left on the cluster which results in a Coulomb energy e2/2R. Due to this presumption, the differences of the relaxation energies between particles having diameters of 2 nm and a 5 nm, respectively, is about 0.15 eV. This difference is within the range of the statistical deviation. The smaller the particles the more significant becomes the influence of the relaxation energies. However, the initial state effect, which is related to a real evolution of the electronic structure of the metal particles from the discrete levels typical for atoms to the bulk band structure, lowers the electron binding energy and can compensate this increasing relaxation energy partially. Simple size effects cannot explain the observed exceptional low binding energy of Au particles supported on TiO2 or SiO2. Therefore, other effects must play a more important role.4.3 Structural arrangement
The effect of structural arrangements of Au atoms in the surface of nanosized particles on the binding energy must be considered, too. The relative number of atoms in surface, edge and corners depends on the particle size.24It is well known, that the Au4f7/2 electrons of surface Au atoms have a binding energy of 83.6 eV (0.4 eV lower than that of bulk atoms), which is explained by the lower coordination number of surface atoms.25 A similar effect, i.e. a dependence of the electronic state on the structural arrangement for small particles was observed by experiments as well as calculations. For small particles below about 2 nm size, where low-coordinated surface atoms and atoms at edges and corners should dominate, a change of the initial states can be expected. This is not the case for particles larger than 3 nm, where the change of the electron binding energy is caused by final state contributions. In experiments generally a positive shift to higher binding energies dominates due to the final state contribution. In an investigation of Cu clusters deposited on ultrathin films of alumina by Wu et al., the separation of the initial state contribution from the relaxation energy was possible.26 For very small clusters a negative initial state contribution up to 0.6 eV was estimated. In a theoretical work, Mottet et al. calculated the local electronic structure of Pd clusters.27 For decreasing coordination number, the binding energies shift to significantly lower values relative to the bulk value.
Beside a dependence of the size of the gold particles on the kind of support, the latter has an influence on the degree of rounding of the particles. Fig. 7 shows representative gold particles of nearly the same sizes on the different supports SiO2, TiO2, ZrO2 and ZnO after a pretreatment in hydrogen at 573 K. On SiO2 and TiO2, the particles are nearly spherical, whereas on the other supports extended facets of the gold surface are visible, in particular for Au/ZnO. On particles exhibiting surfaces with large facets, the amounts of atoms at edges, corners or steps are significantly lower than for rounded particles. Due to this fact, a lower binding energy of the Au4f7/2 electrons originating from the atoms of the nearly spherically-shaped particles on SiO2 and TiO2 supports can be expected, compared to the Au 4f7/2 electron binding energy of the atoms from the gold particles supported on ZrO2 and ZnO.
 | ||
| Fig. 7 Representative gold particles on different supports: upper left: Au/TiO2-DP, upper right: Au/SiO2-I, lower left: Au/ZrO2-DP, lower right: Au/ZnO-I. The gold particles are slightly tilted from ideal zone axes orientation, resulting in visibility of (111) lattice planes only. | ||
This interpretation is strongly supported by the measurements on the system Au–In/ZnO-I. Recently, we could demonstrate, that the addition of indium to the monometallic system Au/ZnO resulted in a selective covering of the facets of the Au particles by the indium species, whereas the corners and edges of the latter remained free of indium. Furthermore, the formation of Au–In intermetallic compounds was not observed.18
The detection of XP spectra is limited to a surface layer of about 2 nm. Due to the geometry of the Au–In particles in the sample Au–In/ZnO-I, the main part of Au 4f electrons analyzed originates from atoms at edges and corners having low coordination numbers. In contrast, for the monometallic sample Au/ZnO-I, the majority of the electrons are ejected from atoms situated in the flat facets of the particles. For Au/ZnO-I, the binding energy of Au 4f electrons is significantly higher, compared to Au–In/ZnO-I (Fig. 6). This is in excellent agreement with the interpretation of the binding energy shift on the basis of the degree of rounding of the gold surface.
It should be noted that the TEM images shown here are measured ex situ. In general, the degree of rounding of particles is further determined by additional interaction with surrounding gases and applied temperatures. Even though this does not influence the comparison of XPS and HRTEM results, the possible effects of temperature and/or high pressure have to be taken into account when catalytic results are correlated with these measurements.
5. Conclusions
The binding energy of an electron as measured by XPS is affected by the coordination number of the respective atom. Due to this observation, a strong influence of the degree of rounding of nanoparticles on the binding energies of the electrons ejected from the atoms forming this particle is well understandable. Different degree of rounding of the Au clusters originated from different particle–support interactions are a possible explanation for the different Au binding energy found for different supports.Moreover, an additional effect of the pretreatment on the properties of the Au binding electrons was observed. One reason for this effect might be an electron transfer from the support to the particle resulting in a charging of the particles. Another possibility explaining this pretreatment effect is a modification of the particle–support interaction leading to changes of the degree of rounding of the Au particles. To solve this problem an investigation with in situ methods might be necessary.
Acknowledgements
C.M. thanks Dr. H. Hofmeister (Max Planck Institute of Microstructure Physics Halle, Germany) for his support in performing electron microscopy. This work has been supported by the Federal Ministry for Science and Education under Grant 03D0028A0. P.C. thanks the Fonds der Chemischen Industrie for financial support.References
- M. Haruta, Catal. Today, 1997, 36, 153 CrossRef CAS.
- C. Mohr, H. Hofmeister, M. Lucas and P. Claus, Chem. Ing. Tech., 1999, 71, 869 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 164 CrossRef CAS.
- S. Minicò, S. Scirè, C. Crisafulli, A. M. Visco and S. Galvagno, Catal. Lett., 1997, 47, 273 Search PubMed.
- D. Guillemot, V. Yu. Borovkov, V. B. Kazansky, M. Polisset-Thfoin and J. Fraissard, J. Chem. Soc., Faraday Trans., 1997, 93, 3587 RSC.
- H. Kageyama, S. Tsubota, K. Kadono, K. Fukumi, T. Akai and M. Haruta, J. Phys. IV, 1997, 7, C2–935 Search PubMed.
- F. Boccuzi, A. Chiorino, M. Manzoli, D. Andreeva and T. Tabakova, J. Catal., 1999, 188, 176 CrossRef.
- M. A. Bollinger and M. A. Vannice, Appl. Catal. B: Environ., 1996, 8, 417 CrossRef CAS.
- L. Guczi, D. Horváth, Z. Pászti, L. Tóth, Z. E. Horváth, A. Karacs and G. Petõ, J. Phys. Chem. B, 2000, 104, 3183 CrossRef CAS.
- F. E. Wagner, S. Galvagno, C. Milone, A. M. Visco, L. Stievano and S. Calogero, J. Chem. Soc., Faraday Trans., 1997, 93, 3403 RSC.
- G. Ertl and J. Küppers, Low Energy Electrons and Surface Chemistry, VCH, Weinheim, 1985 Search PubMed.
- C. R. Henry, Surf. Sci. Rep., 1998, 31, 231 CrossRef.
- J.-D. Grunwaldt, M. Maciejewski, O. S. Becker, P. Fabrizioli and A. Baiker, J. Catal., 1999, 186, 458 CrossRef CAS.
- C. Mohr, H. Hofmeister, M. Lucas and P. Claus, Chem. Eng. Technol., 2000, 23, 324 CrossRef CAS.
- P. Claus, A. Brückner, Ch. Mohr and H. Hofmeister, J. Am. Chem. Soc., 2000, 122, 11430 CrossRef CAS.
- S. Schimpf, M. Lucas, C. Mohr, U. Rodemerck, A. Brueckner, J. Radnik, H. Hofmeister and P. Claus, Catal. Today, 2002, 72, 63 CrossRef CAS.
- O. Böse, E. Kemnitz, A. Lippitz and W. E. S. Unger, Fresenius’ J. Anal. Chem., 1997, 358, 175 CrossRef CAS.
- C. Mohr, H. Hofmeister, J. Radnik and P. Claus, J. Am. Chem. Soc., submitted Search PubMed.
- T. L. Barr, J. Vac. Sci. Technol. A, 1989, 7, 1677 CrossRef CAS.
- J. P. S. Badyal, in Coadsorption, Promoters and Poison, Springer-Verlag, Berlin, 1993, p. 311 Search PubMed.
- A. Sanchez, S. Abbet, U. Heiz, W.-D. Schneider, H. Häkkinen, R. N. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573 CrossRef CAS.
- G. C. Bond, Metal-Support and Metal Additive Effects in Catalysis, ed. B. Imelik, C. Naccache, G. Courier, H. Praliaud, P. Meriaudeau, P. Gallezot, G. A. Martin and J. C. Vedrine, Elsevier, Amsterdam, 1982, ch. 1 Search PubMed.
- A. Zwijnenburg, A. Goossens, W. G. Sloof, M. W. J. Craje, A. M. van der Kraan, L. Jos de Jongh, M. Makkee, J. A. Moulijn, J. Phys. Chem. B, submitted Search PubMed.
- R. van Hardefeld and F. Hartog, Surf. Sci., 1969, 15(2), 189–230 CrossRef CAS.
- P. H. Citrin, G. K. Wertheim and Y. Baer, Phys. Rev. Lett., 1978, 41, 1425 CrossRef CAS.
- Y. Wu, E. Garfunkel and T. E. Madey, J. Vac. Sci. Technol. A, 1996, 14, 1662 CrossRef CAS.
- C. Mottet, G. Treglia and B. Legrand, Surf. Sci., 1996, 675, 352.
| This journal is © the Owner Societies 2003 |