Reversible modification of the absorption properties of single-walled carbon nanotube thin films via nitric acid exposure
Frank
Hennrich
a,
Ralf
Wellmann
b,
Sharali
Malik
a,
Sergei
Lebedkin
a and
Manfred M.
Kappes
ab
aInstitut für Nanotechnologie, Forschungszentrum Karlsruhe, D-76021, Karlsruhe
bInstitut für Physikalische Chemie, Universität Karlsruhe, D-76128, Karlsruhe, Germany
First published on 15th November 2002
Abstract
Exposure of thin films of as-prepared single-walled carbon nanotubes (SWNT) to nitric acid induces dramatic bleaching of the first and second interband transitions of semiconducting tubes (S1 and S2). These near-IR absorption features can be restored by annealing of treated SWNT films to elevated temperatures. Annealing may also be accomplished by visible laser irradiation thus allowing for spatially resolved modifications to SWNT thin film optical absorption.
1. Introduction
A major barrier to the application of single walled carbon nanotubes (SWNTs)1–3 in electronics4 and photonics5 is their as yet still relatively poor materials definition on a molecular scale. All known production methods generate “as-prepared” tube samples manifesting typically poorly quantified contamination by amorphous carbon, fullerenes and catalyst particles–with the latter usually surrounded by shells of chemically resilient turbostratic carbon.6 “Harvesting” and transfer of as-prepared materials generally introduces further contaminants (e.g. adsorbates). In terms of yield of defect free SWNTs, the laser ablation method is arguably the most selective of the available production methods.1 Here, highest yields ranging above 70% of evaporated carbon can be achieved with a nickel/cobalt catalyst, which under optimal conditions generates SWNTs having a narrow diameter distribution between about 1.2 and 1.4 nm. There are thought to be about 30 metallic and semiconducting tubes differing in helicity (and diameter) in this size range.7 Their relative abundances in the Ni/Co material generated are not known in detail. The tubes form in close packed bundles generally 10 to 100 nm in diameter. The level of bundle uniformity with respect to the content of individual tubes as well as the respective defect densities of these tubes remain to be characterized in detail.Often Ni/Co SWNTs are purified in a multistep procedure which first involves refluxing in dilute nitric acid for several days.8 This removes a large fraction of the amorphous carbon and some of the metal alloy particles. Purities of >99% SWNTs by weight have been reported for such materials based upon thermogravimetric analyses (TGA).8 Various experiments, including acid–base titration,9 determination of CO210 released following vacuum heating, FTIR-analysis,11 proton NMR12 and electronic spectroscopy of SWNT derivatives8 have shown that nitric acid workup of as-prepared SWNT material induces the formation of up to 5 atom% carboxylic acid functional groups depending on the extent of acid exposure.
In addition, several recent publications have inferred that nitric acid treatment of SWNT materials also leads to intercalation of HNO3 within tube bundles and consequently to (further) modification of tube electronic properties via hole doping:13 much as in the nitric acid intercalation of graphite.14 Such intercalation has been shown to lead to a measurable expansion of the inter-nanotube spacing.15 Furthermore characteristic changes to the far- and near-IR absorption features of HNO3 purified, Ni/Co catalyst derived laser generated SWNTs have been observed following vacuum heating.13,16 In particular it was noted that the cross section of the first, lowest energy, interband transition in the semiconducting tubes of such samples (S1) is enhanced (by a factor of about 2). This has been attributed to the removal of intercalated NO3− and the associated refilling of the nanotube valence band. In-situ measurements of the effect of introducing NO3− into as-prepared material would be helpful in clarifying the mechanism.
A common difficulty in SWNT materials studies to date is, that the production, purification and sample preparation protocols are not (yet) fully reproducible with respect to detailed spectroscopic features. Consequently, quantitative comparison of different samples is difficult. This is a particular problem if commercial samples are studied whose chemical history is often not known in detail. Here we attempt to resolve the issue of the spectroscopic consequences of nitric acid intercalation by means of absorption spectroscopic probes of thin (∼500 nm) free standing films of various kinds of SWNT samples which we have generated for the purposes of the present study. The thin film sample configuration8 allows more detailed insight into the underlying electronic and vibrational excitations - and thus molecular structure - than has previously been possible. In particular the high material uniformities make it possible to rapidly probe multi-step processes via simple transmission measurements on the same sample.
We show that in particular the van Hove singularities S1 and S2 of as prepared semiconducting tubes can be essentially fully bleached upon short term exposure to nitric acid. Furthermore this effect (on near-IR absorption) is largely reversible if the resulting material is heated under vacuum. This can also be accomplished by laser irradiation at 514 nm. Elemental analysis and IR absorption measurements were used in order to probe materials composition prior and subsequent to annealing. The results show that intercalated (NO3−)(H+) is partially converted to covalently bound –NO2 moieties upon heating. Optical microscopy was used to examine the spatial resolution of laser induced absorption changes for free standing nanotube thin films. Finally we describe similar experiments for nanotube films mounted on a non-absorbing glass substrate.
2. Experimental
2.1 Materials: production and composition
SWNT soots were prepared by laser ablation using a carbon composite rod containing a Ni/Co catalyst (Toyo Tanso, Japan) as has been previously described.17 SWNT soots were treated to generate as prepared and purified buckypapers. We discuss each of the materials encountered in turn.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C pyrolysis in pure O2. Weight percentages are accurate to 1, 3, 5 and 5% of the numbers given, respectively. Cobalt and nickel compositions were determined by atomic emission spectroscopy using standard dissolution methods. Metal compositions are accurate to within 3%. Note that the target rod already contained small amounts of hydrogen and oxygen, presumably deriving from the production process. This appears to be essentially transferred to the nanotube soot as is the overall metal content. The soot is somewhat richer in oxygen than the target rod likely due to metal oxidation and gas adsorption in transfer.
°C pyrolysis in pure O2. Weight percentages are accurate to 1, 3, 5 and 5% of the numbers given, respectively. Cobalt and nickel compositions were determined by atomic emission spectroscopy using standard dissolution methods. Metal compositions are accurate to within 3%. Note that the target rod already contained small amounts of hydrogen and oxygen, presumably deriving from the production process. This appears to be essentially transferred to the nanotube soot as is the overall metal content. The soot is somewhat richer in oxygen than the target rod likely due to metal oxidation and gas adsorption in transfer.
| Material | C | O | N | H | Co | Ni |
|---|---|---|---|---|---|---|
| Carbon rod | 88.3 | 0.8 | 0.034 | 0.8 | 5.4 | 5.4 |
| Raw SWNT soot | 85.8 | 2.0 | <0.5 | 0.8 | 6.7 | 6.6 |
| SWNT (I) | 78.3 | 7.1 | 1.1 | 2.6 | 4.3 | 4.3 |
| SWNT (II) | 65.7 | 25.3 | 1.5 | 2.7 | 1.3 | 1.2 |
000 gn for 10 min). The supernatant liquid was decanted and the solid was again redispersed in DMF for a further purification cycle. Overall four dispersion/centrifugation/decantation cycles were carried out. The decanted liquid contained primarily amorphous carbon and metal particles in DMF suspension (as determined by oxidative thermogravimetric analysis (TGA) after filtering, washing with acetone and drying in vacuum). The residual solid SWNT material was redispersed in water containing 0.2 weight% of the surfactant Triton X-100 (Roth, Germany) in order to obtain as-prepared SWNT “buckypapers” and films by subsequent filtering/washing (see below). This material will be designated SWNT (I) throughout. Elemental analysis of SWNT (I) shows it to contain roughly 80% abundance of carbon by weight. The iterative dispersion/centrifugation/decantation achieves a reduction in metal content from 13 weight% in the raw material to about 8.6 weight% in SWNT (I)
(see Table 1). A corresponding reduction in non-SWNT carbon content is also observed (see TGA measurement: Fig. 1). This is consistent with partial physical separation of nanotube bundles from small carbon coated and uncoated metal catalyst particles. The small nitrogen contents determined in the elemental analysis are likely due to residual adsorbed DMF. Scanning electron microscopy (SEM) analysis of SWNT (I) shows the primary impurities to be amorphous carbon as well as residual (partially oxidized) metal particles. Freshly synthesized cobalt nanoparticles in the 20 nm range are known to be extremely reactive towards oxidation and are coated with one or two monolayers of cobalt oxide when transferred to air.18 Consequently we believe that oxidized metal particles, together with small amounts of residual water and surfactant accounts for the bulk of the oxygen present in SWNT (I) samples. We note in passing that it is possible but less convenient to prepare thin films of SWNT (I) using only DMF without any surfactant–as pointed out in our recent publication (ref. 8).
 | ||
| Fig. 1 Thermogravimetric analysis of the SWNT materials studied here (100 sccm of Ar/O2 at 92 ∶ 8 v/v; 5°C min−1): shown are measurements for SWNTs (I), SWNTs (II) and DMF dispersible residue from DMF purification of raw material (see text). | ||
000 gn before removing the remaining acid solution by decanting. The solid residue was then resuspended in water containing 0.2 weight% of the surfactant Triton X-100 (Roth, Germany). The concentration of SWNTs was about 0.3 mg ml−1. In order to remove remaining small particles, this suspension was run over/through (and flushed with additional Triton X-100 solution) a home-built ultrafiltration cell using 200 nm pore sized membrane filters (Schleicher&Schuell, Germany, cellulose nitrate, 90 mm ∅). This generates residual suspensions of a material (which does not pass through the filter) which we designate as SWNT (II). Solid SWNT (II) samples for elemental analysis were then prepared by filtering the residual suspension and flushing the filtered solid with acetone. The resulting buckypaper comprises approximately 98% by weight of material which can be oxidized to volatiles as determined by TGA, i.e. assuming that residual inorganic ash corresponds to stoichiometric metal oxides. Following literature precedents we had previously equated this 98–99% weight component with total nanotube content (ref. 8). Elemental analysis of SWNT (II) as indicated in Table 1 however shows a significant enhancement in nitrogen content relative to raw SWNT soot due to nitric acid exposure (see below). We will return to this point below.
While metal content is reduced as would be expected for acid treated material, the elemental analysis of SWNT (II) also indicates a significant increase in oxygen and hydrogen relative to SWNT (I). This is due both to the incorporation of oxygen atoms into the nanotubes (e.g. in the form of COOH groups) as well as to the presence of additional surfactant. Note that elemental composition determinations required sample sizes of >60 mg. It was therefore necessary to prepare large amounts of buckypaper, i.e. thick films. Thick papers are difficult to make without significant surfactant content even after repeated solvent washings. The surfactant Triton X-100 in its molecular form does not contain nitrogen. Therefore, the overall elemental composition for SWNT (II) papers is consistent with a nitric acid content of up to 6.75 weight%. As we will argue below, IR spectra in fact suggest the presence of significant amounts of in-tube-bundle intercalated NO3− and possibly small amounts of covalently bound –NO2. Note in comparison, that the corresponding graphite nitrate stage 1 intercalation compound has a stoichiometry of C4.3H0.83NO3, i.e. that the nitric acid loading of SWNT (II) is still comparatively small.
Fig. 1 compares TGA analyses of (i) DMF decantates obtained in generating SWNT (I), (ii) SWNT (I) and (iii) SWNT (II) to a TGA measurement for graphite. Note the reduced inorganic ash percentage for SWNT (II) relative to SWNT (I) as roughly consistent with the elemental analysis. Further structure in the TGA curves obtained for SWNT (I) and SWNT (II) is due to the (oxidation) catalytic activity of the residual metal. The DMF soluble fraction that was washed away by suspension/centrifugation burns at higher temperatures than the SWNTs (I) which is consistent with the inference that this fractions contain a higher portion of amorphous carbon.
2.2 Sample treatments
In all cases, thin film samples were mounted on a stainless steel frame and transferred through air prior to sample treatment or spectroscopic measurement.°C in a furnace under argon (1 bar throughout). Samples were typically retained at elevated temperature for 1 h prior to cooling to room temperature, whereupon the samples were transferred through air and an optical and IR absorption measurement performed. For comparison purposes several samples were iteratively heated, cooled and probed (with anneal temperatures increasing up to 600°C) without visible damage to film morphology.
2.3 Physical characterization
Electronic absorption of thin films was measured under ambient conditions using a UV–vis–NIR spectrophotometer (Carey 5E, Varian) at a resolution of 1 nm. As in a previous study we assume that there are no significant light scattering contributions to the extinction measurements. Spectra were recorded over the wavelength range 3300–200 nm using a sample area of 0.3 cm2. No significant changes to absorption features were observed in repetitive spectroscopic scans over the same sample area indicating that the light fluences used for electronic absorption measurements were low enough to leave samples unchanged. In some cases measurements were analysed by subtracting absorption contributions due to the SWNT π-plasmon. For this, 100 points of the plasmon background were fit to a spline, and smoothed by hand.IR transmission measurements (Bruker IFS 66v) over the range 400–4000 cm−1 were carried out at room temperature under vacuum (1 mbar) at a resolution of 2 cm−1. For further data analysis, thin film transmission was converted to absorbance and background plasmon absorption was subtracted as described above for UV–visible–NIR measurements.
In addition to spectroscopic measurements on free-standing films mounted as described above, we also studied the microscopic outcome of laser annealing (using a 1 mm slit to irradiate only part of the film). After exposure, films were detached from their frames and subsequently floated onto glass surfaces by the procedure previously described.8 Following drying under ambient conditions, such samples were probed using optical microscopy as well as scanning electron microscopy (SEM). Preliminary irradiation experiments were also performed on films which had been surface mounted prior to irradiation. Significantly higher laser fluences were necessary in order to produce comparable spectroscopic changes, depending in detail on thermal contact to the support. This will be reported in more detail in a future publication.
3. Results and discussion
3.1 Optical absorption overview
 | ||
| Fig. 2 Far-IR to UV spectra of “reference materials” SWNT (I) and (II): Absorbance versus wavenumber (on a logarithmic scale). | ||
°C, the lowest anneal temperature studied. Note that with increasing anneal temperature/anneal cycles, the optical absorption properties of thermally annealed SWNT (II) films approach but do not quite reach those of SWNT (I) samples: S1, S2 and M features respectively saturate at about 50% of the corresponding cross sections of SWNT (I) samples. Furthermore, bands are somewhat broader and the positions of the absorption centroids shift slightly to lower energy, suggestive of changes to the tube diameter distributions (→larger tubes) in going from SWNT (I) to SWNT (II) consistent with faster etching of smaller diameter tubes as has been pointed out by several authors.26
 | ||
| Fig. 3 Optical absorption measurements for an SWNT (II) film after annealing to various temperatures under argon as described in text, in comparison to untreated SWNT (I) and the same SWNT(II) thin film at room temperature prior to heating. (a) Absorbance versus wavenumber (logarithmic scale; full spectral range) and (b) relative absorbance (background subtracted) versus photon energy (linear scale); spectra for each temperature are offset for clarity in (b). Annealing-temperature-dependent measurements were performed in sequence of increasing temperature. | ||
 | ||
| Fig. 4 (a) Optical absorption spectra for an SWNT (II) film subject to reversible S1 bleaching in the temporal order: pristine film, after exposure to HNO3 and after subsequent laser anneal (see text for details). (b) Absorption spectra for an SWNT (II) film before and after laser anneal (also shown is the corresponding absorption spectrum for a pristine SWNT (I) film: dotted line). | ||
°C. The absorption properties of untreated SWNT (I) films remained unchanged upon equivalent irradiation. A more accurate approximation of the effective temperatures achieved upon laser irradiation requires measurements of the thermal conductivity of thin films. These are not yet available.
Analogous experiments were also performed by exposing a free standing SWNT (II) film to laser irradiation through a 1 mm metallic slit. While scanning electron microscopy does not show obvious structural changes, optical microscopy using a white light source allows easy recognition of the boundary region: the irradiated surface appears significantly darker. The boundary is characterized by network structures reminiscent of individual intertwined tube bundles presumably indicating pathways of preferential thermal conduction. The microscopy allows us to set an upper limit to the spatial resolution of such laser induced absorption changes: approximately 1 μm.
3.2 FTIR measurements
The overview spectra shown in Fig. 2 for both reference materials contain two broad IR absorption features: a strong far IR absorption with a maximum below the instrumental range and the near-IR S1 transition. Superimposed on these bands, which have been extensively discussed in a previous study13 and which for the purposes of the following discussion we will regard as background, are various weak vibrational features.SWNT (I) films show primarily two sets of vibrational features, one broad set of very weak absorptions centred near 1400 cm−1 and a second sharp set of ca. two absorptions near 3000 cm−1. The latter are due to adsorbed hydrocarbon impurities: primarily surfactant and pump oil derived. Note that in a previous study we have presented surfactant free measurements of SWNT (I) thin films generated using only DMF which showed no absorption cross section in these spectral ranges.
Untreated SWNT (II) absorption measurements also manifest two sets of vibrational features. Again we observe the sharp C–H stretch region absorptions assigned to the surfactant–as seen in SWNT (I) films. However, the weak lower frequency vibrations that are observed in SWNT (I) films are obscured by a much stronger set of absorptions with two local maxima at 1630 and 1762 cm−1. These are shown after background subtraction in Fig. 5. The band at 1762 cm−1 is believed to derive from the COOH group. Several authors have reported IR-bands between 1709–1740 as arising from treating SWNTs with concentrated and/or diluted nitric acid and/or mixtures of nitric acid with sulfuric acid10,27–34 and have assigned these to corresponding carboxyl group vibrations.
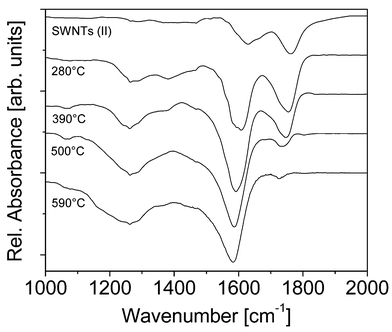 | ||
| Fig. 5 Background subtracted IR absorption measurements for an SWNT (II) thin film annealed to various temperatures in argon: see text for details. Shown are relative absorptions for the 1000–2000 cm−1 spectral range after background subtraction. The data sets are shown offset to each other for clarity. They indicate loss of –COOH and partial conversion of intercalated NO3− to covalently bound NO2. | ||
Several effects are observed in the SWNT (II) IR spectra upon annealing at increasing temperatures: (i) the C–H stretch vibrations rapidly diminish in intensity, (ii) the intensity of the C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O stretch vibration of COOH appears first to increase slightly upon heating to 280°C and then decreases during further heating; at the same time it shifts from 1762 to 1728 cm−1, (iii) the twin NO3− features at 1660 and 1630 cm−1 are replaced by a broad absorption at 1602 cm−1 after first heating to 280°C; this feature shifts to 1581 cm−1 during further heating and (iv) a new peak appears in the 3400 cm−1 region (see Fig. 3b). We attribute the first two effects to desorption of surfactant/hydrocarbons and decomposition of COOH groups with concomitant evolution of CO and CO2. Note that the latter gases have been seen in a number of TPD studies on related nanotube materials at similar temperatures.
O stretch vibration of COOH appears first to increase slightly upon heating to 280°C and then decreases during further heating; at the same time it shifts from 1762 to 1728 cm−1, (iii) the twin NO3− features at 1660 and 1630 cm−1 are replaced by a broad absorption at 1602 cm−1 after first heating to 280°C; this feature shifts to 1581 cm−1 during further heating and (iv) a new peak appears in the 3400 cm−1 region (see Fig. 3b). We attribute the first two effects to desorption of surfactant/hydrocarbons and decomposition of COOH groups with concomitant evolution of CO and CO2. Note that the latter gases have been seen in a number of TPD studies on related nanotube materials at similar temperatures.
The third observation is consistent with the loss of NO3− together with the formation of covalently bound –NO2 we assign the new spectral features to unresolved symmetric and antisymmetric stretch vibrations of the latter. We speculate that the increase in the 3400 cm−1 absorption cross section is either due to the formation of covalently bound –OH or is indicative of tube coalescence. Several studies on unpurified, raw SWNTs show that diameter doubling can occur upon heating to temperatures near 1400°C,35,36 under electron irradiation in a transmission electron microscope with simultaneous heating to 800°C37 and upon heating in H2.38 Perhaps (acid generated) defects can catalyze the process at lower temperatures in analogy to what has been observed for fullerenes.39
3.3 Consequences and conclusions
Our observations show that free standing thin films of as-prepared laser vaporized SWNTs having diameters between 1.2 and 1.4 nm may be easily hole doped by exposing them to nitric acid. This results in intercalation of HNO3 within the tube bundles and a dramatic reduction in the absorption cross section of the S1 transition in the near-IR spectral range. Heating such films to temperatures in excess of 300°C, either within an oven or by laser irradiation leads to partial conversion of intercalated NO3− to covalently bound NO2. Correspondingly, we observe an almost complete restoration of the optical properties of the as-prepared film. Optical microscopy of laser annealed samples suggests that it is possible to induce such absorption changes locally. Preliminary measurements for glass mounted SWNT thin films indicate that such “write–erase” cycles are practicable with lateral resolutions below 1 μm. Thus it should be possible to fabricate strongly absorbing patterns with length scales in the range of S1 transition wavelengths.
Acknowledgements
This research was supported by the Deutsche Forschungsgemeinschaft under Sonderforschungsbereich 551. We also gratefully acknowledge support by BMBF.References
- A. Thess, R. Lee, P. Nikolaev, H. Dai, P. Petit, J. Robert, C. Xu, H. Lee, S. G. Kim, D. T. Colbert, G. Scuseria, D. Tomanek, J. E. Fischer and R. E. Smalley, Science, 1996, 273, 483 CAS.
- S. Iijima and T. Ichihashi, Nature, 1991, 363, 603 CrossRef.
- P. M. Ajayan, J. M. Lambert, P. Bernier, L. Barbedette, C. Colliex and J. M. Planeix, Chem. Phys. Lett., 1993, 215, 509 CrossRef CAS.
- S. J. Wind, J. Appenzeller, R. Martel, V. Derycke and P. Avouris, Appl. Phys. Lett., 2002, 80, 3817 CrossRef CAS.
- M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593 CrossRef CAS.
- M. Monthioux, B. W. Smith, B. Burteaux, A. Claye, J. E. Fischer and D. E. Luzzi, Carbon, 2001, 39, 1251 CrossRef CAS.
- H. Kuzmany, B. Burger, M. Hulman, J. Kürti, A. G. Rinzler and R. E. Smalley, Europhys Lett., 1998, 44, 518 Search PubMed.
- F. Hennrich, S. Lebedkin, S. Malik, J. Tracy, M. Barczewski, H. Rösner and M. Kappes, Phys. Chem. Chem. Phys., 2002, 4, 2273 RSC.
- H. Hu, P. Bhowmik, B. Zhao, M. A. Hamon, M. E. Itkis and R. C. Haddon, Chem. Phys. Lett., 2001, 345, 25 CrossRef CAS.
- D. B. Mawhinney, V. Naumenko, A. Kuznetsova, J. T. Yates, Jr., J. Liu and R. E. Smalley, Chem. Phys. Lett., 2000, 324, 213 CrossRef.
- M. A. Hamon, H. Hu, P. Bhowmik, S. Niyogi, B. Zhao, M. E. Itkis and R. C. Haddon, Chem. Phys. Lett., 2001, 347, 8 CrossRef CAS.
- C. Goze Bac, P. Bernier, S. Latil, V. Jourdain, A. Rubio, S. H. Jhang, S. W. Lee, Y. W. Park, M. Holzinger and A. Hirsch, Curr. Appl. Phys., 2001, 1, 149 Search PubMed.
- M. E. Itkis, S. Niyogi, M. E. Meng, M. A. Hamon, H. Hu and R. C. Haddon, Nano Lett., 2002, 2, 155 CrossRef CAS.
- M. P. Conrad and H. L. Strauss, Phys. Rev. B, 1985, 31, 6669 CrossRef CAS.
- C. Bower, A. Kleinhammes, Y. Wu and O. Zhou, Chem. Phys. Lett., 1998, 288, 481 CrossRef CAS.
- Z. H. Yu and L. E. Brus, J. Phys. Chem. A, 2000, 104, 10995 CAS.
- S. Lebedkin, P. Schweiss, B. Renker, S. Malik, F. Hennrich, M. Neumaier, C. Stoermer and M. M. Kappes, Carbon, 2002, 40, 417 CrossRef CAS.
- D. P. Dinega and M. G. Bawendi, Angew. Chemie., Int. Ed., 1999, 38, 1788 Search PubMed.
- H. Kataura, Y. Kumazawa, I. Umezu, S. Suzuki, Y. Ohtsuka and Y. Achiba, Synth. Met., 1999, 103, 2555 CrossRef CAS.
- J. W. G. Wildoer, L. C. Venema, A. G. Rinzler, R. E. Smalley and C. Dekker, Nature, 1998, 391, 59 CrossRef CAS.
- T. W. Odom, J.-L. Huang, P. Kim and C. M. Lieber, Nature, 1998, 391, 62 CrossRef CAS.
- M. Ichida, S. Mizuno, Y. Tani, Y. Saito and A. Nakamura, J. Phys. Soc. Jpn., 1999, 68, 3131 Search PubMed.
- P. Petit, C. Mathis, C. Journet and P. Bernier, Chem. Phys. Lett., 1999, 305, 370 CrossRef CAS.
- O. Jost, A. A. Gorbunov, W. Pompe, T. Pichler, R. Friedlein, M. Knupfer, M. Reibold, H.-D. Bauer, L. Dunsch, M. S. Golden and J. Fink, App. Phys. Lett., 1999, 75, 2217 Search PubMed.
- C. T. White and J. W. Mintmire, Nature, 1998, 394, 29 CrossRef CAS.
- A. Koshio, M. Yudasaka and S. Iijima, Chem. Phys. Lett., 2001, 341, 461 CrossRef CAS.
- B. N. Khare, M. Meyyappan, A. M. Cassell, C. V. Nguyen and J. Han, Nano Lett., 2002, 2, 73 CrossRef CAS.
- A. Kuznetsova, D. B. Mawhinney, V. Naumenko, J. T. Yates, Jr., J. Liu and R. E. Smalley, Chem. Phys. Lett., 2000, 321, 292 CrossRef CAS.
- A. Kuznetsova, I. Popova, J. T. Yates, Jr., M. J. Bronikowski, C. B. Huffman, J. Liu, R. E. Smalley, H. H. Hwu and J. G. Chen, J. Am. Chem. Soc., 2001, 123, 10699 CrossRef CAS.
- M. S. P. Shaffer, X. Fan and A. H. Windle, Carbon, 1998, 36, 1603 CrossRef.
- J. Chen, A. M. Rao, S. Lyuksyutov, M. E. Itkis, M. A. Hamon, H. Hu, R. W. Cohn, P. C. Eklund, D. T. Colbert, R. E. Smalley and R. C. Haddon, J. Phys. Chem. B, 2001, 105, 2525 CrossRef CAS.
- J. Chen, M. A. Hamon, H. Hu, Y. Chen, A. M. Rao, P. C. Eklund and R. C. Haddon, Science, 1998, 282, 95 CrossRef CAS.
- E. V. Basiuk, V. A. Basiuk, J.-G. Banuelos, J.-M. Saniger-Blesa, V. A. Pokrovskiy, T. Yu. Gromovoy, A. V. Mischanchuk and B. G. Mischanchuk, J. Phys. Chem. B, 2002, 106, 1588 CrossRef CAS.
- A. Kukovecz, Ch. Kramberger, M. Holzinger, H. Kuzmany, J. Schalko, M. Mannsberger and A. Hirsch, J. Phys. Chem. B, 2002, 106, 6374 CrossRef CAS.
- P. Nikolaev, A. Thess, A. G. Rinzler, D. T. Colbert and R. E. Smalley, Chem. Phys. Lett., 1997, 266, 422 CrossRef CAS.
- M. Yudasaka, H. Kataura, T. Ichihashi, L.-C. Qin, S. Kar and S. Iijima, Nano Lett., 2001, 1, 487 CrossRef CAS.
- M. Terrones, H. Terrones, F. Banhart, J.-C. Charlier and P. M. Ajayan, Science, 2000, 288, 1226 CrossRef CAS.
- S. L. Fang, A. M. Rao, P. C. Eklund, P. Nikolaev, A. G. Rinzler and R. E. Smalley, J. Mater. Res., 1998, 13, 2405 CAS.
- R. D. Beck, C. Stoermer, C. Schulz, R. Michel, P. Weis, G. Bräuchle and M. M. Kappes, J. Chem. Phys., 1994, 101, 3243 CrossRef CAS.
| This journal is © the Owner Societies 2003 |