A new strategy for folding oligo(m-phenylene ethynylenes)†
Xiaowu
Yang
a,
Amy L.
Brown
a,
Mako
Furukawa
b,
Shoujian
Li
c,
Wendy E.
Gardinier
a,
Eric J.
Bukowski
a,
Frank V.
Bright
a,
Chong
Zheng
c,
Xiao Cheng
Zeng
b and
Bing
Gong
*a
aDepartment of Chemistry, University at Buffalo, The State University of New York, Buffalo, New York, 14260, USA. E-mail: bgong@chem.buffalo.edu; Fax: (+1) 716 645 6963; Tel: (+1) 716 645 6800
bDepartment of Chemistry, University of Nebraska-Lincoln, Lincoln, Nebraska 68588, USA
cDepartment of Chemistry, Northern Illinois University, DeKalb, Illinois 60115, USA
First published on 20th November 2002
Abstract
Backbone-rigidified oligo(m-phenylene ethynylenes) fold into crescent or helical conformations in non-polar organic solvents.
Helical structures are ubiquitously found in Nature1 and have inspired current efforts in developing unnatural oligomers and polymers that fold into well-defined conformations.2–18 In spite of the progress made so far, the foldamer field is still in its infancy. For example, the generation of well defined cavities, a feature usually seen at the tertiary and quaternary structural levels of biopolymers, has been realized in few unnatural foldamer systems.6,18 Moore et al. have developed an elegant sytem of folding oligo(m-phenylene ethynylenes) (m-PE) in polar organic solvents based on a solvophobically driven mechanism. We describe here a different strategy for folding oligo(m-PEs) in non-polar organic solvents. Our strategy involves backbone-rigidification by H-bonding as shown by the general structure 1. Depending on chain length, crescent and helical conformations are obtained.
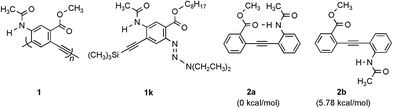
It is known that a small barrier (∼0.6 kcal mol−1) exists for the internal rotation of diphenylacetylene 2.19 The conformations of simple o- and m-PE oligomers and polymers should thus be very flexible and random. By incorporating an intramolecular H-bond into 2, the resulting 2a should adopt a well-defined conformation enforced by this additional non-covalent interaction. Ab initio molecular orbital calculations20 indicated that 2a adopted a completely planar conformation that was rigidified by its intramolecular H-bond. Deviation from the planar conformation of 2a by interrupting the intramolecular H-bond led to a rapid increase in energy. A rotational barrier of 7.19 kcal mol−1 between conformers 2a and 2b was also found.
Crystals of dimer 2c were obtained from ethyl acetate by slow cooling and the X-ray structure is shown in Fig. 1.‡20 The intramolecular H-bond, as expected, leads to a planar conformation that is consistent with the above calculation.
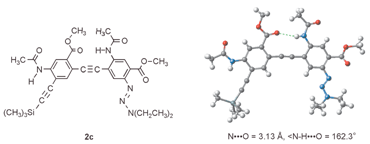 | ||
| Fig. 1 The crystal structure of 2c. | ||
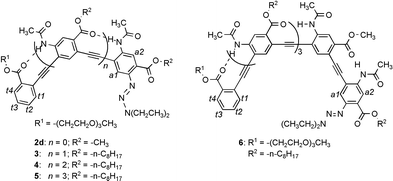
If the same intramolecular H-bond is introduced into PE oligomers of various chain lengths, well-defined conformations may be enforced. Thus, oligomers 2c–d, 3, 4, 5 and 6 with two, three, four, five and six benzene rings, respectively, along with monomer 1k, were examined by 1H NMR in CDCl3 (500 MHz).
The chemical shift values of amide 1H signals indicated the formation of intramolecular H-bonds: the spectrum of 1k (2 mM), which can not form any intramolecular H-bond, showed an NH signal at 7.92 ppm. In contrast, one of the NH signals of dimer 2c (2 mM) moved significantly downfield (9.22 ppm), and the other appeared at 8.03 ppm, which indicated that one was involved in intramolecular H-bonding and the other was not. For oligomers 2d and 3–6 (2 mM), all NH signals appeared at significantly downfield positions (8.98 to 9.46 ppm), consistent with their involvement in intramolecular H-bonding.
Upon diluting a sample of tetramer 4 from 8 mM to 0.063 mM, the three NH signals of 4 showed minuscule shifts of 0.008, 0.016 and 0.015 ppm, confirming the existence of intramolecular H-bonding.
Variable temperature (VT) 1H NMR study of the amide signals of tetramer 4 provided additional evidence for the prevalence of intramolecular H-bonds. At 2 mM and from −20 to 60 °C in CDCl3, the three amide signals of 4 showed small upfield shifts (−2.3 × 10−3, −2.2 × 10−3 and −2.2 × 10−3 ppm K−1) reminiscent of intramolecular H-bonding.17,21 Instead of moving upfield, the amide resonances of hexamer 6 (2 mM) showed very small downfield shifts (2–4 × 10−3 ppm K−1) with increasing temperature (−20 to 60 °C in CDCl3).20 This observation, along with the fact that the amide proton signals of 6 appeared at positions upfield to those of 2d–5, suggests that (1) the amide protons of 6 werer involved in intramolecular H-bonding, and (2) at 2 mM, hexamer 6 was, to certain extent, involved in stacking interaction that was disrupted at elevated temperatures.
Comparing the chemical shifts of aromatic protons t1–t4 and a1,a2 on the end residues of oligomers 2d–6 revealed an interesting trend:20 from dimer 2d to pentamer 5, the chemical shifts of protons a1,a2 and t1–t4 showed very small changes. In contrast, protons t1–t4 and a1,a2 of hexamer 6 all showed obvious upfield shifts. The shifts are particularly significant (up to 0.5 ppm) for protons t2 and t3. A similar trend of upfield shifts was observed for the ethyl protons of the diethyltriazenyl group: while those of 2d–5 remained constant, those of 6 showed upfield shifts for up to 0.7 ppm. These results can only be explained by the corresponding oligomers adopting a curved backbone: while oligomers 2d–5 are not long enough, hexamer 6 reaches a length that allows its two termini to be brought into close proximity, which caused the observed upfield shifts.22
If hexamer 6 adopts a folded conformation, the chemical shifts of its end proton signals should be sensitive to change in temperature. The folded conformation will be partially interrupted with increasing temperature, which should change (increase) the distance between the two ends and should thus cause the 1H NMR signals of the end protons to move downfield. This was indeed the case. The NMR signals of protons t2 and t3 showed obvious downfield shifts with rising temperature, protons t1 and t4 were less sensitive. In contrast, the corresponding protons t2 and t3 of tetramer 4 showed very small temperature-dependent changes in their chemical shifts.20
Tetramer 4 was then examined by two-dimensional (NOESY) 1H NMR studies (Fig. 2). NOEs clearly indicated the side chain contacts between protons aMe, bMe and cMe of the acetamido methyl groups and the α-CH2 groups of the ester side chains. NOEs between the protons of the amide NH groups and the ester α-CH2 groups were also observed. The observed NOEs were consistent with a crescent conformation enforced by the intramolecular H-bonds.
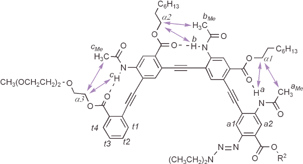 | ||
| Fig. 2 NOEs between adjacent amide and ester groups of tetramer 4 as revealed by NOESY (8 mM in CDCl3, 500 MHz, 263 K, mixing time: 0.3 s).20 | ||
The NOESY spectrum of hexamer 6 also revealed numerous NOEs corresponding to side chain-side chain contacts.20 One significant difference was observed between 6 and 4 (Fig. 2): the NOESy spectrum of 6 revealed NOEs between proton t1 (7.36 ppm), and those of the two diethyltriazenyl methyl groups, Me1 and Me2 (0.79 and 1.10 ppm). On the other hand, similar NOEs involving protons t1 and Me1 and Me2 were absent in the spectrum of 4. The observed end-to-end contacts for hexamer 6, combined with the chemical shift changes, are fully consistent with the helical conformation shown in Fig. 3. Modeling shows that such a folded helical conformation has a hydrophobic cavity of ∼8 Å across.
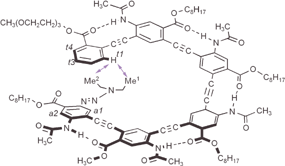 | ||
| Fig. 3 End-to-end NOE contacts between proton t1 and the methyl protons Me1 and Me2 of hexamer 6 as revealed by NOESY (8 mM in CDCl3, 500 MHz, 263 K, mixing time: 0.3 s).20 | ||
The UV spectra of 2d–620 revealed chain length-dependent features. All five compounds showed a very strong absorption band at ∼330 nm. In chloroform (2 μM), the spectra of 2d, 3 and 4 were very similar. However, a new band appeared for pentamer 5 at ∼370 nm and much more so for hexamer 6. The 370-nm shoulder for 5 or 6 should not be due to intermolecular aggregation because nearly identical spectra for 2d–6 were obtained at a higher concentration (10 μM) in chloroform.20 In methanol/chloroform (1∶1), the 370-nm bands of 5 and 6 greatly diminished, while the spectra of 2d–4 remained unchanged. Except for dimer 2d, the longer oligomers were all highly fluorescent. The highest energy emission feature lay between 420 and 440 nm. New emission features appeared for pentamer 5 (525 nm, not very obvious) and hexamer 6 (530 nm, very obvious). These new bands are mostly likely due to the intramolecular exciton coupling between the two ends of 5 or 6.
This study has demonstrated the feasibility of designing PE oligomers with stably folded conformations based on backbone-rigidification. By incorporating building blocks with the two ethynyl linkages being placed in a para-geometry on the same bezene ring, the curvature of the backbones can be adjusted. This, combined with the localized nature of backbone-rigidification, allows the development of PE helices with larger interior cavities.
The NASA and the NIH are acknowledged for funding. Part of the computational work was done on the University of Nebraska Research Computing Facilities computer.
Notes and references
- D. S. Lawrence, T. Jiang and M. Levett, Chem. Rev., 1995, 95, 2229 CrossRef CAS.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS.
- D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 13071 CrossRef CAS.
- D. Seebach, M. Overhand, F. N. M. Kühnle, B. Martinoni, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1996, 79, 913 CrossRef CAS.
- L. A. Cuccia, J.-M. Lehn, J.-C. Homo and M. Schmutz, Angew. Chem., Int. Ed., 2000, 39, 233 CrossRef CAS.
- C. W. Wu, T. J. Sanborn, K. Huang, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 6778 CrossRef CAS.
- Y. Hamuro, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1997, 119, 10587 CrossRef CAS.
- R. S. Lokey and B. L. Iverson, Nature, 1995, 375, 303 CrossRef CAS.
- M. Hagihara, N. J. Anthony, T. J. Stout, J. Clardy and S. L. Shreiber, J. Am. Chem. Soc., 1992, 114, 6568 CrossRef CAS.
- A. B. Smith III, T. P. Keenan, R. C. Holcomb, P. A. Sprengeler, M. C. Guzman, J. L. Wood, P. J. Carroll and R. Hirschmann, J. Am. Chem. Soc., 1992, 114, 10672 CrossRef.
- D. Yang, J. Qu, B. Li, F. F. Ng, X.-C. Wang, K.-K. Cheung, D.-D. Wang and Y.-D. Wu, J. Am. Chem. Soc., 1999, 121, 589 CrossRef CAS.
- V. Berl, I. Huc, R. G. Khoury, R. G. Krische and J.-M. Lehn, Nature, 2000, 407, 720 CrossRef CAS.
- A. Taratani, T. S. Hughes and J. S. Moore, Angew. Chem., Int. Ed., 2002, 41, 325 CrossRef.
- B. Gong, H. Q. Zeng and J. Zhu, Proc. Natl. Acad. Sci. USA, 2002, 99, 11583 CrossRef CAS.
- J. Zhu, R. D. Parra, H. Q. Zeng, E. Skrzypczak-Jankun, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2000, 122, 4219 CrossRef CAS.
- B. Gong, Chem. Eur. J, 2001, 7, 4336 CrossRef CAS.
- R. D. Parra, H. Q. Zeng, J. Zhu, C. Zheng, X. C. Zeng and B. Gong, Chem. Eur. J., 2001, 7,, 4352 CrossRef CAS.
- K. Okuyama, T. Hasegawa, M. Ito and N. Mikami, J. Phys. Chem., 1984, 88, 1711 CrossRef CAS.
- See ESI† for details.
- B. Gong, Y. Yan, H. Q. Zeng, E. Skrzypczak-Jankunn, Y. W. Kim, J. Zhu and H. A. Ickes, J. Am. Chem. Soc., 1999, 121, 5607 CrossRef CAS.
- S. J. Perkins, Biol. Magn. Reson., 1982, 4, 193 Search PubMed.
Footnotes |
| † Electronic supplementary information available: experimental data. See http://www.rsc.org/suppdata/cc/b2/b209809a/ |
‡ Crystal data for 2c: C31H37N5O6, M = 603.75, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 9.8811(19), b = 13.014(3), c = 14.189(3) Å, α = 115.143(3), β = 90.863(4), γ = 99.193(4)°, U = 1623.4(5) Å3, Z = 2, μ(Mo-Kα) = 0.121 mm−1, 7198 reflections measured (4472 unique, Rint = 0.0222). The final wR(F2) was 0.1907 (all data). CCDC 195111. See http://www.rsc.org/suppdata/cc/b2/b209809a/ for crystallographic data in CIF or other electronic format. , a = 9.8811(19), b = 13.014(3), c = 14.189(3) Å, α = 115.143(3), β = 90.863(4), γ = 99.193(4)°, U = 1623.4(5) Å3, Z = 2, μ(Mo-Kα) = 0.121 mm−1, 7198 reflections measured (4472 unique, Rint = 0.0222). The final wR(F2) was 0.1907 (all data). CCDC 195111. See http://www.rsc.org/suppdata/cc/b2/b209809a/ for crystallographic data in CIF or other electronic format. |
| This journal is © The Royal Society of Chemistry 2003 |