B(C6F5)3-catalyzed formation of B–P bonds by dehydrocoupling of phosphine–boranes†
Jean-Marc
Denis
*a,
Henrietta
Forintos
a,
Helga
Szelke
a,
Loic
Toupet
c,
Thi-Nhàn
Pham
b,
Pierre-Jean
Madec
b and
Annie-Claude
Gaumont
*b
aUniversité de Rennes I, CNRS-UMR 6510, Campus de Beaulieu, F35042, Rennes, France. E-mail: jean-marc.denis@univ-rennes1.fr.
bUniversité de Caen-ISMRA, LCLT, CNRS-UMR 6507, 6 Bd. du Mal Juin, F14050, Caen, France
cUniversité de Rennes, CNRS-UMR 6625, Campus de Beaulieu, F35042, Rennes, France
First published on 25th November 2002
Abstract
Tris(pentafluorophenyl)borane was used as a new catalyst in the formation of P–B bonds by dehydrocoupling of phosphine–boranes.
Catalytic heterodehydrocoupling of covalent hydrides represents an interesting route to the formation of element–element bonds. In this way, formation of Si–N and Si–C bonds has been achieved some years ago using group 4 metallocenes as catalysts.1 The Manners group has recently developed a new route to B–P2 and B–N3 bond formation involving catalytic dehydrocoupling of the corresponding phosphine– and amine–boranes with Rh(I) as catalyst. In this preliminary communication we report two selected examples of the use of tris(pentafluorophenyl)borane,4‡ B(C6F5)3 (BX3) as a catalyst for the formation of B–P bonds under mild conditions by dehydrocoupling of phosphine–borane adducts. Our results will be compared with those obtained with the transition-metal catalyzed reactions.2 A mechanism will be proposed.
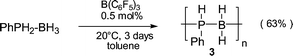
Dehydrogenative coupling of H3B·PPhH21 has recently been performed by refluxing a toluene solution overnight in the presence of 0.5 mol% Rh(I) as catalyst.2 High molecular weight poly(phosphinoboranes) [PhPH–BH2]n were thus obtained (Table 1, entry 1). We observed the dehydro-condensation of 1 at 20 °C in a toluene solution containing 0.5 mol% of B(C6F5)3§ (entry 2).¶ Evolution of gas could be observed during the first 2 h. The reaction was completed after 3 days (yield 63% in isolated products). 31P- and 11B-NMR displayed two separated groups of signals (≈1∶1 ratio). The 31P NMR (toluene) presented a broad signal at δ −48.9 (d, JPH ≈ 348 Hz) and poorly resolved peaks from δ −52 to −56, respectively. The corresponding 11B NMR spectra revealed a braod peak at δ −35.5 and a shoulder at δ −33.2. All these data are characteristic of four-coordinated boron centres attached to two phosphorus.2 Similar results were obtained by performing the reaction at 90 °C for 3 h (entry 2). The 31P and 11B values observed at δ −48.9 and −35.8, respectively, are consistent with the formation of poly(phenylphosphino)boranes 3 recently described in the literature.2 The poorly resolved 31P signals from δ −52 to −56,2 in the PH region suggested the presence of low molecular-weight oligomeric or cyclic structures. The coexistence of two main fractions corresponding roughly to Mw = 3900 and 830 were observed by size exclusion chromatography measurements (according to polystyrene standards) with a polydispersity index of 2.3 and 1.9, respectively. Differential scanning calorimetry confimed these results and indicated a crystalline structure for 3 (Tm = 215 and 194 °C, respectively). Poly(phenylphosphinoboranes) 3 are air- and moisture-stable in the solid state.
| Entry | Starting material | Catalyst (reagent) | Conditions | Conv.a (%) | Product(s) |
31P NMR
δ (JPH) |
11B NMR
δ |
|---|---|---|---|---|---|---|---|
| a Progress of the reaction was monitored by 31P NMR. b See ref. 2. c Shoulder. | |||||||
| 1 | 1 b |
RhI
(0.5 mol%) |
110 °C, 15 h
(toluene) |
100 | 3 |
−48.9 (360)
|
−34.7 |
| 2 | 1 |
(C6F5)3B
(0.5 mol%) |
20 °C, 3 days (toluene)
or 90 °C, 3 h (toluene) |
100 | 3 |
−48.9 (348)
−52 to −56 |
−35.8
−33c |
| 3 | 2 |
(C6F5)3B
(5 mol%) |
Bubbling PH3 and BH3 into CH2Cl2/(C6F5)3B
then 70 °C overnight |
100 |
H3P(BH2PH2)nBH3
oligomers |
−104 (t, 362)
−109 (t, 342) −115 (q, 356) |
Main peaks at
−32 and −35 |
| 4 | 2 |
(C6F5)3B
(5 mol%) |
Bubbling PH3 and BH3 into
CH2Cl2/(C6F5)3B then 90 °C for 24 h |
100 | [PH2BH2]n4 |
Broad peak
centred at −107 |
Broad peak
centred at −32 |
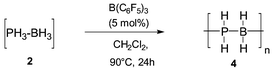
The main problem encountered in extending the dehydrocoupling to the complex H3B·PH32 was due the weakness of the P–B bond and its fast dissociation at temperatures as low as −30 °C under atmospheric pressure.5 We used for this reaction safety equipment which allowed to bubble at −50 °C PH3(g)6 and B2H6(g)7 into a CH2Cl2 solution of B(C6F5)3 (≈5 mol% with regard to BH3) and thereafter to heat the solution to the desired temperature. Oligomerisation started around +20 °C and was completed by heating at 70 °C overnight (entry 3). The oligomeric structure H3P(BH2PH2)nBH3 was proposed from the 31P and 11B spectra [disappearance of the peak at δP −119 corresponding to the complex 2 and formation of three broad peaks at δ −104 (t, JPH 362 Hz), −109 (t, JPH 342) (PH2 groups) and a small peak at δ −115 (q, JPH 356) (terminal PH3 group)]. The 11B NMR showed complex resonances (main peaks at δ −32 and −35 in good agreement with such a structure).2 The material obtained by heating the solution at 90 °C for one day (entry 4) was assigned to 4 on the basis of the 31P NMR (very broad peak from δ −95 to −120) and 11B NMR (very broad peak centred at δ −32). The white solid material thus obtained after evacuation of the solvent was very sensitive to air and moisture. Very fast oxidation prevented reliable elemental and HRMS analyses.
These results prove the efficiency of B(C6F5)3 as a new catalyst for the preparation of poly(phosphinoboranes) by dehydrocoupling of phosphine–boranes (Table 1). In order to get a better understanding of the mechanism, a stoechiometric experiment between 5||** and 1 was performed. Attempts to isolate the primary product 7 resulting from dehydrocoupling reaction were unsuccessful, even at −10 °C. The poly(phosphinoborane) 3 was in all the cases the main observed product.
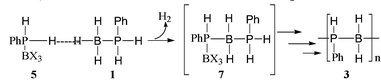
We supppose from this experiment that complex 5 (Fig. 1)†† formed by ligand exchange was probably the reactive intermediate in the catalytic dehydrocoupling. The acceptor strength of Lewis-acidic perfluororinated triarylborane compounds is well established.8,9 This effect should contribute to activate the P–H bond by withdrawing electron density from the phosphorus, making dehydrocoupling easier. Polymerisation presumably followed a process involving iterative dehydrocoupling reactions and BX3/BH3 exchanges.
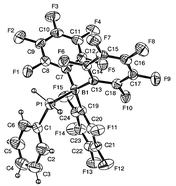 | ||
| Fig. 1 Molecular structure of 5. Selected bond length (Å): P(1)–B (1) 2.039. | ||
Poly(phosphinoboranes) 3, 4 were also formed by another route involving in the first step the formation of the complexes 8, 9 (Fig. 2)†† respectively and slow decomposition of these intermediates (20 °C for 8 and 110 °C, 3 h for 9) To explain the formation of the complex BX3–SMe210, a transient three-coordinate complex R(H)P–BH2 was suggested as intermediate.10
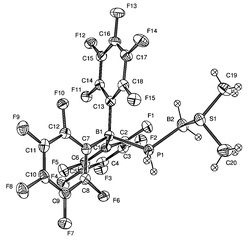 | ||
| Fig. 2 Molecular structure of 9. Selected bond length (Å): P(1)–B(1) 2.049. | ||
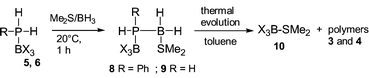
In conclusion, we have presented a new and efficient route to poly(phosphinoboranes) by using the strong Lewis acid B(C6F5)3 as catalyst. The ease of the dehydrocoupling of 1 and 2 is attributed to the strong acidic character of the P–H bond of complexes 5 and 6. The polymerisation presumably followed a process involving iterative dehydrocoupling reactions and BX3/BH3 exchanges. This work addresses mechanistic questions. Extension of this dehydrocoupling route to other element–element bond formation is in progress.
Notes and references
- For a review: F. Gauvin, J. F. Harrod and H. G. Woo, Adv. Organomet. Chem., 1998, 42, 463 Search PubMed.
- H. Dorn, R. A. Singh, J. A. Massey, J. M. Nelson, C. A. Jaska, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2000, 122, 6669 CrossRef CAS.
- C. A. Jaska, K. Temple, A. J. Lough and I. Manners, Chem. Commun., 2001, 962 RSC.
- Reviews: (a) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345–354 RSC; (b) K. Ishihara and H. Yamamoto, Eur. J. Chem., 1999, 527–538 Search PubMed.
- H. Schmidbaur, T. Wimmer, J. Lachmann and G. Muller, Chem. Ber., 1991, 124, 275 CAS.
- D. Semenzin, G. Etemad-Moghadam, D. Albouy and M. Koenig, Tetrahedron Lett., 1994, 35, 3297 CrossRef CAS.
- J. V. B. Kanth and H. C. Brown, Inorg. Chem., 2000, 39, 1795 CrossRef.
- D. C. Bradley, I. S. Harding, A. D. Keefe, M. Motevalli and D. H. Zheng, J. Chem. Soc., Dalton Trans., 1996, 3931 RSC.
- D. C. Bradley, M. B. Hursthouse, M. Motevalli and D. H. Zheng, J. Chem. Soc., Chem. Commun., 1991, 7 RSC.
- Review: P. P. Power, Angew. Chem., Int. Ed . Engl., 1990, 29, 449 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental section. See http://www.rsc.org/suppdata/cc/b2/b206559b/ |
| ‡ B(C6F5)3 is a Lewis acid of comparable strength to BF3. Application of this water-tolerant reagent as a catalyst in organic synthesis is rapidly growing.4 |
| § The use of anhydrous grade B(C6F5)3 was critical. For each experiment, the product was sublimed by plunging the flask maintained under vacuum (0.02 mbar) into an oil-bath previously heated at 105 °C. All the manipulations should be carried out under neutral gas in dry solvents and reagents. |
| ¶ Typical experiment: 2.5 × 10−4 mol of the borane complex PhPH2·BH3 in toluene (400 μL) was slowly added into a toluene solution (100 μL) of the freshly sublimated BX3 (6.0 × 10−4 g; 1.2 × 10−6 mol, 0.5 mol%) and the solution was maintained at the considered temperature. Progress of the reaction was monitored by 31P NMR. Traces of free phosphine or phosphine oxide were sometimes detected by 31P. |
| || An authentic sample of 5 was easily prepared and fully characterised by 11B and 1H NMR, HRMS, and single crystal X-ray diffraction. |
| ** An authentic sample of 10 was easily prepared and characterised by NMR and X-ray diffraction. CCDC 189752. (ESI†. |
†† Crystal data for 5: C24H7BF15P, M = 622.8, T
= 293(2) K, λ
= 0.71069, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , unit cell dimensions: a = 8.070(5), b = 11.337(9), c = 12.992(9) Å, α = 86.66(9) β = 77.22(7), γ = 87.560(8)°, V = 1156.7(14) Å3, Z = 2, Dc
= 1.786 g cm−3, μ
= 0.254 mm−1, F(000) = 612, crystal size: 0.32 × 0.24 × 0.16 mm, θ Range for data collection 1.61–24.97°, index range, 0 < h < 9, −13 < k < 13, −15 < 1 < 15, reflections collected: 4389, independent reflections: 4070 [Rint = 0.0156], reflections observed (>2σ): 2495, refinement method, full-matrix least-squares on F2, data/restraints/parameters, 4070/0/377, goodness-on-fit on F2
= 1.009, final R indices [(I > 2σ(I)]: R1 = 0.0422, wR2 = 0.0788, R indices (all data): R = 0.0989, wR = 0.0918, largest diff. peak and hole, 0.196 and −0.209 e Å−3. CCDC 189751.Crystal data for 9: C20H10B2F15PS, M = 619.93, T
= 293(2) K, λ
= 0.71069, triclinic, space group P, unit cell dimensions: a = 9.6926(4), b = 10.6562(5), c = 12.3789(7) Å, α = 64.353(2) β = 86.081(2), γ = 84.137(3)°, V = 1146.16(10) Å3, Z = 2, Dc
= 1.796 g cm−3, μ
= 0.343 mm−1, F(000) = 612, crystal size: 0.12 × 0.10 × 0.03 mm, θ Range for data collection 1.83–27.61°, index range, 0 < h < 12, − 13 < k < 13, −15 < 1 < 16, reflections collected: 5270, reflections observed: 5270, goodness of fit: 1.029, final R indices: R = 0.0478, wR = 0.1320. CCDC 189752.See http://www.rsc.org/suppdata/cc/b2/b206559b/ for crystallographic data in CIF or other electronic format. , unit cell dimensions: a = 8.070(5), b = 11.337(9), c = 12.992(9) Å, α = 86.66(9) β = 77.22(7), γ = 87.560(8)°, V = 1156.7(14) Å3, Z = 2, Dc
= 1.786 g cm−3, μ
= 0.254 mm−1, F(000) = 612, crystal size: 0.32 × 0.24 × 0.16 mm, θ Range for data collection 1.61–24.97°, index range, 0 < h < 9, −13 < k < 13, −15 < 1 < 15, reflections collected: 4389, independent reflections: 4070 [Rint = 0.0156], reflections observed (>2σ): 2495, refinement method, full-matrix least-squares on F2, data/restraints/parameters, 4070/0/377, goodness-on-fit on F2
= 1.009, final R indices [(I > 2σ(I)]: R1 = 0.0422, wR2 = 0.0788, R indices (all data): R = 0.0989, wR = 0.0918, largest diff. peak and hole, 0.196 and −0.209 e Å−3. CCDC 189751.Crystal data for 9: C20H10B2F15PS, M = 619.93, T
= 293(2) K, λ
= 0.71069, triclinic, space group P, unit cell dimensions: a = 9.6926(4), b = 10.6562(5), c = 12.3789(7) Å, α = 64.353(2) β = 86.081(2), γ = 84.137(3)°, V = 1146.16(10) Å3, Z = 2, Dc
= 1.796 g cm−3, μ
= 0.343 mm−1, F(000) = 612, crystal size: 0.12 × 0.10 × 0.03 mm, θ Range for data collection 1.83–27.61°, index range, 0 < h < 12, − 13 < k < 13, −15 < 1 < 16, reflections collected: 5270, reflections observed: 5270, goodness of fit: 1.029, final R indices: R = 0.0478, wR = 0.1320. CCDC 189752.See http://www.rsc.org/suppdata/cc/b2/b206559b/ for crystallographic data in CIF or other electronic format. |
| This journal is © The Royal Society of Chemistry 2003 |