Computational evidence that the inverse kinetic isotope effect for reductive elimination of methane from a tungstenocene methyl–hydride complex is associated with the inverse equilibrium isotope effect for formation of a σ-complex intermediate†
Kevin E.
Janak
,
David G.
Churchill
and
Gerard
Parkin
*
Department of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: parkin@chem.columbia.edu; Fax: 212 932 1289; Tel: 212 854 8247
First published on 28th November 2002
Abstract
Calculations on [H2Si(C5H4)2]W(Me)H demonstrate that the interconversion between [H2Si(C5H4)2]W(Me)H and the σ-complex [H2Si(C5H4)2]W(σ-HMe) is characterized by normal kinetic isotope effects for both reductive coupling and oxidative cleavage; the equilibrium isotope effect, however, is inverse and is the origin of the inverse kinetic isotope effect for the overall reductive elimination of methane.
Transition metal compounds in which a hydrocarbon is coordinated to a metal by a 3-center–2-electron M⋯H–C interaction, i.e. so-called [M](σ-HR) σ-complexes, are recognized to be important intermediates in the oxidative addition and reductive elimination of C–H bonds.1 In addition to low temperature spectroscopic studies, the principal evidence for σ-complex intermediates is derived from (i) the observation of deuterium exchange between hydride and alkyl sites, e.g. [M](CH3)D → [M](CH2D)H, and (ii) the measurement of kinetic isotope effects (KIEs).2 For example, because a single step reaction is almost invariably characterized by a normal primary KIE (i.e. kH/kD > 1), the observation of an inverse KIE (i.e. kH/kD < 1) for reductive elimination of alkane from [M](CH3)H and [M](CD3)D isotopologues is commonly taken to imply the existence of an intermediate in a multistep reaction prior to the rate determining step (Scheme 1).2 Specifically, the observed KIE for reductive elimination (kre) of an alkane reflects a composite of the effect of deuterium substitution on the individual rate constants for (i) reductive coupling (krc) to form the σ-complex, (ii) oxidative cleavage (koc) to regenerate the alkyl–hydride complex, and (iii) alkane dissociation (kd). Depending upon which step is rate determining and the magnitude of the individual isotope effects, the overall isotope effect for reductive elimination may be either normal or inverse. Although there are many reports of KIEs for overall reductive elimination of alkane, there is a paucity of data concerned with the isotope effects for each of the individual fundamental transformations. Therefore, in this paper we employ computational methods to determine kinetic and equilibrium isotope effects pertaining to the reductive elimination of methane.
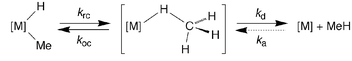 | ||
| Scheme 1 | ||
Previous studies on Cp2W(Me)H,3 Cp*2W(Me)H4 and [Me2Si(C5Me4)2]W(Me)H5 and their d4-isotopologues have demonstrated that the reductive elimination of methane is characterized by an inverse kinetic isotope effect, thus implying the existence of a σ-complex intermediate prior to rate determining loss of methane. Of these complexes, the reductive elimination is most inverse for the ansa-complex [Me2Si(C5Me4)2]W(Me)H, with a value of 0.45 at 100 °C, thereby making it the most appropriate candidate for dissecting the origin of the overall kinetic isotope effect. Hence, we performed a series of DFT (B3LYP) calculations pertaining to reductive elimination of methane from [Me2Si(C5Me4)2]W(Me)H.6,7 A sequence of linear transit geometry optimizations that progressively couple the CMe–H bond resulted in the generation of the σ-complex intermediate [Me2Si(C5Me4)2]W(σ-HMe) via a {[Me2Si(C5Me4)2]W(σ-HMe)}‡ transition state (Fig. 1). Subsequent dissociation of methane from the σ-complex [Me2Si(C5Me4)2]W(σ-HMe) generates the tungstenocene {[Me2Si(C5Me4)2]W} intermediate. However, since the 16-electron intermediate is calculated to be 12.9 kcal mol−1 more stable as a triplet than as a singlet, it is evident that dissociation of methane from [Me2Si(C5Me4)2]W(σ-HMe) involves a spin crossover.7a,b The structure of [Me2Si(C5Me4)2]W(σ-HMe) at the crossing point corresponds to the transition state for dissociation on the enthalpy surface and was determined by a procedure analogous to that reported for [H2C(C5H4)2]W(σ-HMe).7a
![Calculated enthalpy surface for reductive elimination of CH4 from [Me2Si(C5Me4)2]W(Me)H.](/image/article/2003/CC/b209684f/b209684f-f1.gif) | ||
| Fig. 1 Calculated enthalpy surface for reductive elimination of CH4 from [Me2Si(C5Me4)2]W(Me)H. | ||
The computation of isotope effects requires knowledge of the vibrational frequencies of the participating species. Frequency calculations are, however, highly computationally intensive. Therefore, it was necessary to perform such studies on a computationally simpler system in which the methyl groups of the [Me2Si(C5Me4)2] ligand are replaced by hydrogen atoms. This simplification considerably facilitates the calculation, while still retaining the critical features of the molecules of interest.
Kinetic isotope effects are conventionally determined by the expression: KIE = kH/kD = SYM·MMI·EXC·ZPE, where SYM is the symmetry factor, MMI is the mass moment of inertia term, EXC is the excitation term and ZPE is the zero point energy term.8,9 Calculated primary and secondary KIE values for the individual transformations pertaining to the overall reductive elimination of methane from [H2Si(C5H4)2]W(Me)H are summarized in Table 1, illustrating several important points. Firstly, the primary KIE for reductive coupling of [H2Si(C5H4)2]W(Me)X (X = H, D) to give the σ-complex [H2Si(C5H4)2]W(σ-XMe) is small, but normal (1.05). Likewise, the microscopic reverse, i.e. oxidative cleavage of [H2Si(C5H4)2]W(σ-XMe), is also normal (1.60). The equilibrium isotope effect (EIE) for the interconversion of [H2Si(C5H4)2]W(Me)X and [H2Si(C5H4)2]W(σ-XMe), however, is inverse (0.65), a consequence of the fact that the KIE for oxidative cleavage is greater than that for reductive coupling.10 Secondary isotope effects do not play a significant role, with values close to unity for the interconversion of [H2Si(C5H4)2]W(CX3)H and [H2Si(C5H4)2]W(σ-HCX3): krc(H)/krc(D) = 1.02, koc(H)/koc(D) = 1.09, and Kσ(H)/Kσ(D) = 0.94. Analysis of the individual SYM, MMI, EXC and ZPE terms indicates that it is the zero point energy term that effectively determines the magnitude of the isotope effects for the interconversion of [H2Si(C5H4)2]W(Me)H and [H2Si(C5H4)2]W(σ-HMe).
| SYM | MMI | EXC | ZPE | IE | ||
|---|---|---|---|---|---|---|
| k rc(H)/krc(D) |
p
s p&s |
1
1 1 |
1.00
1.00 1.00 |
1.05
0.98 1.04 |
1.00
1.04 1.00 |
1.05
1.02 1.04 |
| k oc(H)/koc(D) |
p
s p&s |
1
1 1 |
1.01
1.00 1.00 |
1.03
1.05 1.08 |
1.54
1.04 1.60 |
1.60
1.09 1.73 |
| K σ(H)/Kσ(D) |
p
s p&s |
1
1 1 |
0.99
1.00 0.99 |
1.01
0.94 0.96 |
0.65
1.00 0.63 |
0.65
0.94 0.60 |
| k d(H)/kd(D) |
p
s p&s |
1
1 1 |
1.00
0.98 0.98 |
0.90
0.92 0.85 |
0.98
1.23 1.15 |
0.88
1.11 0.96 |
| k re(H)/kre(D) |
p
s p&s |
1
1 1 |
1.00
0.98 0.98 |
0.91
0.86 0.82 |
0.63
1.23 0.72 |
0.58
1.04 0.58 |
| K d(H)/Kd(D) |
p
s p&s |
0.25
0.25 1 |
0.69
0.36 0.28 |
1.14
1.80 2.00 |
0.94
1.43 1.25 |
0.19
0.24 0.69 |
The KIE for dissociation of methane from a σ-complex has been postulated to be small.3 Dissociation of methane from [H2Si(C5H4)2]W(σ-HMe) would likewise be expected to exhibit a small KIE, especially since the C–H bond in the σ-complex is almost fully formed (dC–H = 1.17 Å). Despite the complication that the transition state for dissociation occurs at the singlet–triplet crossing point,11 frequency calculations on singlet [H2Si(C5H4)2]W(σ-HMe) with the geometry of the crossing point demonstrate that the KIEs for dissociation of methane are indeed close to unity (Table 1). Interestingly, and in contrast to the neglible KIEs, the EIEs for dissociation of methane are large and inverse due to the SYM and MMI terms; in particular, the large inverse MMI term for dissociation of methane from [H2Si(C5H4)2]W(σ-HCH3) and [H2Si(C5H4)2]W(σ-DCD3) is a consequence of the fact that isotopic substitution has a substantial effect on the moments of inertia of a molecule as small as methane.12
By predicting both a normal kinetic isotope effect for the reductive coupling step and an inverse kinetic isotope effect for the overall reductive elimination, the calculated isotope effects for reductive elimination of methane from [H2Si(C5H4)2]W(Me)H are in accord with the experimental study on [Me2Si(C5Me4)2]W(Me)H.5 For example, the calculated inverse KIE for reductive elimination of methane from [H2Si(C5H4)2]W(CH3)H and [H2Si(C5H4)2]W(CD3)D (0.58)13 compares favorably with the experimental value for [Me2Si(C5Me4)2]W(CH3)H and [Me2Si(C5Me4)2]W(CD3)D (0.45).5 Analysis of the isotope effects for the various steps provides conclusive evidence that the principal factor responsible for the inverse nature of the KIE for the overall reductive elimination is the inverse equilibrium isotope effect for the interconversion of [H2Si(C5H4)2]W(Me)H and [H2Si(C5H4)2]W(σ-HMe). The calculations therefore reinforce the notion that inverse primary kinetic isotope effects for reductive elimination of alkanes imply the existence of a σ-complex intermediate prior to rate determining loss of alkane.
It is important to emphasize that while the majority of R–H versus R–D reductive elimination reactions are characterized by inverse KIEs, there are several examples where a normal KIE is observed, e.g. (R3P)2Pt(Me)H.2 For these examples, it is the reductive coupling step that is postulated to be rate determining.4 Thus, regardless of whether the overall reductive elimination of alkane is characterized by a normal or inverse KIE, the reductive coupling exhibits a normal KIE.14
In summary, calculations on [H2Si(C5H4)2]W(Me)H provide the first theoretical evidence that the inverse kinetic isotope effect for reductive elimination of methane is a manifestation of the existence of a σ-complex intermediate. Specifically, the inverse kinetic isotope effect for reductive elimination is a consequence of an inverse equilibrium isotope effect for interconversion of [H2Si(C5H4)2]W(Me)H and [H2Si(C5H4)2]W(σ-HMe).
We thank the U. S. Department of Energy, Office of Basic Energy Sciences (DE-FG02-93ER14339) for support of this research and Drs Bruce Bender and Mu-Hyun Baik for helpful comments.
Notes and references
- (a) R. H. Crabtree, J. Chem. Soc., Dalton Trans., 2001, 2437 RSC; (b) C. Hall and R. N. Perutz, Chem. Rev., 1996, 96, 3125 CrossRef CAS.
- For recent reviews, see: (a) W. D. Jones, Acc. Chem. Res. Search PubMed in press; (b) R. M. Bullock and B. R. Bender, ‘Isotope Methods in Homogeneous Catalysis’ in Encyclopedia of Catalysis, ed. I. T. Horváth, Wiley, New York, 2003 Search PubMed.
- R. M. Bullock, C. E. L. Headford, K. M. Hennessy, S. E. Kegley and J. R. Norton, J. Am. Chem. Soc., 1989, 111, 3897 CrossRef CAS.
- G. Parkin and J. E. Bercaw, Organometallics, 1989, 8, 1172 CrossRef CAS.
- D. G. Churchill, K. E. Janak, J. S. Wittenberg and G. Parkin, J. Am. Chem. Soc. Search PubMed , in press.
- DFT B3LYP (Jaguar 4.1): Geometry optimizations and frequency calculations (6-31G** and LACVP** basis sets); single point calculations (cc-pVTZ(-f) and LACV3P** basis sets).
- For other calculations pertaining to (CpR)2M(Me)H (M = Mo,W) derivatives, see: (a) J. C. Green and C. N. Jardine, J. Chem. Soc., Dalton Trans., 1998, 1057 RSC; (b) J. C. Green, J. N. Harvey and R. Poli, J. Chem. Soc., Dalton Trans., 2002, 1861 RSC; (c) M.-D. Su and S.-Y. Chu, J. Phys. Chem. A, 2001, 105, 3591 CrossRef CAS.
- B. K. Carpenter, Determination of Organic Reaction Mechanisms, Wiley-Interscience, New York, 1984 Search PubMed.
- We have also determined the isotope effects using (i) the modification that employs the Redlich–Teller product rule, i.e. KIE = SYM·VP·EXC·ZPE, and (ii) the thermodynamic values obtained directly from the DFT calculations. Significantly, the three methods yield very similar results, thereby providing an indication of the reliability of the calculations. See supporting information for comparison of the three methods..
- Jones′ seminal experimental study of [TpMe2]Rh(L)(Me)H has also demonstrated that the inverse EIE for reductive coupling is a result of the normal kinetic isotope effect for reductive coupling being smaller than the normal kinetic isotope effect for oxidative coupling. See: T. O. Northcutt, D. D. Wick, A. J. Vetter and W. D. Jones, J. Am. Chem. Soc., 2001, 123, 7257 Search PubMed.
- As such, the derived transition state is not a well defined stationary point on the enthalpy surface; nevertheless, we have also calculated frequencies at other points on the singlet dissociation surface and find similar values.
- Although there are no experimentally determined EIE values for dissociation of methane from σ-complexes (or association of methane), related values have been reported for other alkanes. Interestingly, both normal and inverse values have been observed. See, for example: (a) S. Geftakis and G. E. Ball, J. Am. Chem. Soc., 1998, 120, 9953 CrossRef CAS; (b) A. A. Bengali, R. H. Schultz, C. B. Moore and R. G. Bergman, J. Am. Chem. Soc., 1994, 116, 9585 CrossRef CAS.
- These calculations assume the preequilibrium approximation (koc ≫ kd) for reductive elimination (kre), thereby corresponding to the most extreme inverse value for the system.
- An inverse KIE for reductive coupling in [Tp]Pt(CH3)H2 has been reported.14a,b However, the validity of this claim has been questioned. See refs. 2(a) and 5. (a) H. C. Lo, A. Haskel, M. Kapon and E. Keinan, J. Am. Chem. Soc., 2002, 124, 3226 CrossRef CAS; (b) M. A. Iron, H. C. Lo, J. M. L. Martin and E. Keinan, J. Am. Chem. Soc., 2002, 124, 7041 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: computational details. See http://www.rsc.org/suppdata/cc/b2/b209684f/ |
| This journal is © The Royal Society of Chemistry 2003 |