Nonlinear amplification of circular dichroism activity upon cyclodimerization of a chiral saddle-shaped porphyrin†
Yukitami
Mizuno
and
Takuzo
Aida
*
Department of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-8656, Japan
First published on 25th November 2002
Abstract
A cyclic dimer of chiral saddle-shaped porphyrin with p-xylylene linkers, upon interaction with mandelic acid, showed an enhanced circular dichroism activity, which was more than 7 times as large as that of a monomeric reference.
Supramolecular induction of molecular chirality has attracted great attention1 in relation to chirality sensing and chiroptical memory devices. We have reported the first example of a chirality-memory molecule2,3 on the basis of a saddle-shaped, fully substituted porphyrin.4 For example, porphyrin 1 bearing two different aryl groups at the adjacent meso-positions is chiral with a symmetry group D2, due to the saddle-shaped conformation (Scheme 1). Although the enantiomers of 1 are hardly separable because of a thermal inversion of the saddle, 1 turns optically active via the interaction with a chiral carboxylic acid such as mandelic acid (MA), since the resulting mixture has a strong energetic bais toward either of the two diastereoisomers.2 Furthermore, 1 remains optically active even when dissolved in acetic acid (achiral), which replaces the interacting chiral acid (MA). Thus, 1 can memorize chiral acid configurations. X-Ray crystallography has shown that 1 forms a hydrogen-bonded complex with two molecules of MA, where the four pyrrole units are pointing up and down alternately.3 Due to this nonplaner structure, the meso-aryl groups are twisted in a clockwise or anticlockwise fashion with respect to the porphyrin macrocycle. The structural feature of the mandelate complex prompted us to explore the interesting possibility that this nonplanar chirality can be translated into a helical chirality when 1 is cofacially linked via appropriate spacers with another molecule of 1. Here we report that a cyclic dimer of 1 with p-xylylene linkers (2) (see ESI†), upon interaction with MA, shows a highly enhanced circular dichroism activity, due to an intramolecular interaction between the two large, chiral chromophore units.
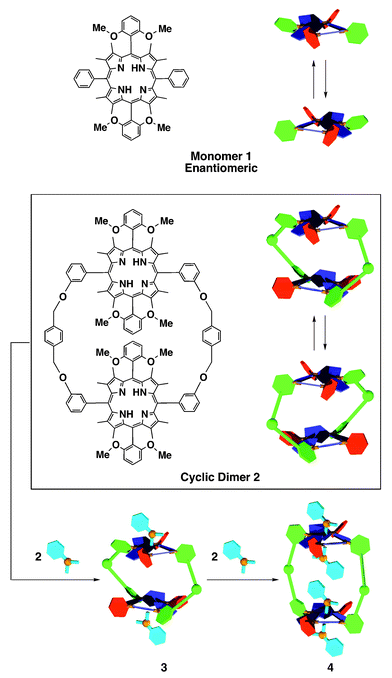 | ||
| Scheme 1 Schematic structures of 1 and 2, and a proposed mechanism for the stepwise binding of mandelic acid with cyclic dimer 2. | ||
Fig. 1 shows a molecular model of 2, created based on the crystal structure of the bis(mandelate) complex of 1,3 where the entire molecule of 2 can be twisted in a helical manner when the absolute configurations of the two saddle-shaped porphyrin units are identical to one another. The molecular model also indicates that 2 possesses only a small inner space, to which guest carboxylic acids are not freely accessible. Cyclic dimer 2 in 1,2-dichloroethane (DCE) showed absorption bands at 440, 542 and 685 nm, which are slightly blue-shifted from those of monomer 1 (444, 543 and 688 nm), indicating a weak electronic interaction between the two cofacial porphyrin units. In contrast with 1, 2 showed a rather broad and complicated 1H NMR spectrum, possibly due to its constrained conformational change as well as multiple ring-current effects of the porphyrin units.
 | ||
| Fig. 1 A molecular model of cyclic dimer 2, based on the X-ray crystallographic structure of the bis(mandelate) complex of monomeric reference 1. | ||
Fig. 2 shows the absorption spectral changes of (a) cyclic dimer 2 and (b) monomeric reference 1, upon titration with (S)-mandelic acid ((S)-MA) in DCE at 23 °C. Although these two spectral change profiles appear to be very similar to one another, Fig. 2(a), at a closer look, suggests a stepwise binding of 2 with (S)-MA (Scheme 1). At molar ratios [(S)-MA]/[2] of 0–2, the spectral change in the Soret region (blue curves) showed an increase in intensity of the absorption band at 467 nm at the expense of the absorption at 440 nm, displaying an isosbestic point at 458 nm. On the other hand, when the molar ratio [(S)-MA]/[2] exceeded 2, the isosbestic point shifted to 452 nm (red curves). This is in contrast with the titration profile of monomeric reference 1 (Fig. 2(b)), where only a single isosbestic point was observed at 458 nm irrespective of the molar ratio of (S)-MA to 1. Thus, it is likely that cyclic dimer 2, at the molar ratio [(S)-MA]/[2] from 0 to 2, interacts with (S)-MA preferentially at the exterior binding sites to form a mono-hydrogen bonded complex (3), and then the third and fourth (S)-MA molecules, upon further addition of (S)-MA, are hydrogen-bonded with 3 from its inner space to form bis-hydrogen-bonded complex 4 (Scheme 1). Although the 1H NMR spectrum (see ESI†) of 2 in the presence of MA was again broad and little informative of this stepwise binding, it showed an upfield shift of the MA signals (e.g., Δδ = −1.7 ppm for Ar-o-H at [(S)-MA]/[2] = 2.8) characteristic of the hydrogen-bonded MA with saddle-shaped porphyrins (ring-current effect).2
![Absorption spectral changes of (a) cyclic dimer 2 (3.9 × 10−6 M) and (b) monomeric reference 1 (3.8 × 10−6 M) upon titration with (S)-mandelic acid ((S)-MA) in 1,2-dichloroethane (DCE) at 23 °C. Blue and red curves are spectra at [(S)-MA]/[host] in ranges of 0–2 and 2–7, respectively.](/image/article/2003/CC/b209789c/b209789c-f2.gif) | ||
| Fig. 2 Absorption spectral changes of (a) cyclic dimer 2 (3.9 × 10−6 M) and (b) monomeric reference 1 (3.8 × 10−6 M) upon titration with (S)-mandelic acid ((S)-MA) in 1,2-dichloroethane (DCE) at 23 °C. Blue and red curves are spectra at [(S)-MA]/[host] in ranges of 0–2 and 2–7, respectively. | ||
Similarly to monomeric reference 1, cyclic dimer 2 became optically active upon mixing with (R)- or (S)-MA. Furthermore, 2 remained optically active when the above mixture was dissolved in acetic acid (see ESI†) where the lifetime of this chirality memory was as long as that of 1. Fig. 3(a) shows the circular dichroism (CD) spectral change of 2 (8.6 × 10−6 M) at 23 °C upon titration with (S)-MA in DCE. Possibly due to the aforementioned stepwise binding (Scheme 1), the spectral change profile is not as simple as that of monomeric reference 1. When the molar ratio [(S)-MA]/[2] was increased up to 2, the CD bands at 435 and 467 nm were both enhanced monotonically (blue curves). On the other hand, when the ratio [(S)-MA]/[2] was further increased, the intensities of these two CD bands became smaller, while new CD bands appeared at 450 and 484 nm (red curves). For clarity, the difference in CD intensities at 467 and 435 nm (Δεmaximum − Δεminimum) of 2 and that of reference 1, plotted against [(S)-MA]/[host], are shown in Fig. 3(b) and (c), respectively, where two contrasting behaviors of 1 and 2 are noted: (1) The value Δεmaximum – Δεminimum of 1 is increased linearly with the amount of (S)-MA, and simply reaches a plateau at the molar ratio [(S)-MA]/[1] of 2 (Fig. 3(c)). (2) The highest value of Δεmaximum − Δεminimum, observed for dimer 2 (260 dm3 mol−1 cm−1) at [(S)-MA]/[2] = 2 ([(S)-MA]/[porphyrin] = 1), is not double but more than 7 times as large as that of monomer 1. Such a large CD enhancement can be partly ascribed to the helical twisting of the entire molecule of 2 (Fig. 1). As already described, the two saddle-shaped porphyrin units of 2 at [(S)-MA]/[2] = 2 are most likely mono-hydrogen-bonded with (S)-MA (3, Scheme 1), and their absolute configurations are identical to one another. Consequently, 2 is forced to adopt a helical twisting either in a clockwise or anticlockwise manner. On the other hand, when the molar ratio [(S)-MA]/[2] exceeds 2, the third and fourth MA molecules start to interact with the inner binding sites of 3 to form 4 (Scheme 1). This inclusion event certainly requires a large expansion of the inner space of the host macrocycle, which is surely uncomfortable for the helical conformation of the molecule. Consequently, the optical activity of the supramolecular complex should be considerably decreased (red curves in Fig. 3(a)).
![(a) Circular dichroism (CD) spectral change of cyclic dimer 2 (3.9 × 10−6 M) upon titration with (S)-mandelic acid ((S)-MA) in 1,2-dichloroethane (DCE) at 23 °C. Blue and red curves are spectra at [(S)-MA]/[host] in ranges of 0–2 and 2–7, respectively. Plots of Δεmaximum
−
Δεminimum of the CD bands of (b) 2 (3.9 × 10−6 M) and (c) 1 (3.8 × 10−6 M) in the Soret region again [(S)-MA]/[host] in DCE at 23 °C.](/image/article/2003/CC/b209789c/b209789c-f3.gif) | ||
| Fig. 3 (a) Circular dichroism (CD) spectral change of cyclic dimer 2 (3.9 × 10−6 M) upon titration with (S)-mandelic acid ((S)-MA) in 1,2-dichloroethane (DCE) at 23 °C. Blue and red curves are spectra at [(S)-MA]/[host] in ranges of 0–2 and 2–7, respectively. Plots of Δεmaximum − Δεminimum of the CD bands of (b) 2 (3.9 × 10−6 M) and (c) 1 (3.8 × 10−6 M) in the Soret region again [(S)-MA]/[host] in DCE at 23 °C. | ||
In conclusion, through spectroscopic studies on the interaction of mandelic acid (MA) with the cyclic dimer of chiral saddle-shaped porphyrin (2) and its monomeric reference (1), we found that the circular dichroism activity is significantly enhanced by a factor of more than 7. Such a nonlinear CD enhancement is most likely due to the translation of the nonplanar chirality of the saddle parts into the helical chirality of the entire molecule. Thus, 2 serves as a powerful chiral sensor, which can read out absolute structures of interacting guest molecules and transcribe them into highly amplified CD signals in the visible region. Considering that the 1∶2 complex of 2 and MA (3, Scheme 1) has a guest binding chiral space between the two saddle-shaped porphyrin units, the supramolecular chirality of cyclic host 2, induced by chiral carboxylic acids, may be utilized for asymmetric recognition and catalysis.
Notes and references
- Selected examples: E. Yashima, T. Matsushima and Y. Okamoto, J. Am. Chem. Soc., 1997, 119, 6345 Search PubMed; T. Mizutani, T. Kurahashi, T. Murakami, N. Matsumi and H. Ogoshi, J. Am. Chem. Soc., 1997, 119, 8991 CrossRef CAS; X. Huang, H. B. Rickman, B. Borhan, N. Berova and N. Nakanishi, J. Am. Chem. Soc., 1998, 120, 6185 CrossRef; M. Takeuchi, T. Imada and S. Shinkai, Angew. Chem., Int. Ed., 1998, 37, 2096 CrossRef CAS; E. Yashima, K. Maeda and Y. Okamoto, Nature, 1999, 399, 449 CrossRef CAS; J. H. K. Ky Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167 CrossRef CAS; L. J. Prins, F. D. Jong, P. Timmerman and D. N. Reinhoudt, Nature, 2000, 408, 181 CrossRef CAS; J. M. Rivera, S. I. Craig, T. Martin and J. Rebek Jr., Angew. Chem., Int. Ed., 2000, 39, 2130 CrossRef CAS; A. Hori, A. Akasaka, K. Biradha, S. Sakamoto, K. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3269 CrossRef CAS.
- Y. Furusho, T. Kimura, Y. Mizuno and T. Aida, J. Am. Chem. Soc., 1997, 119, 5267 CrossRef CAS.
- Y. Mizuno, T. Aida and K. Yamaguchi, J. Am. Chem. Soc., 2000, 122, 5278 CrossRef CAS.
- Selected examples: K. M. Barkigia, M. D. Berber, J. Fajer, C. J. Medforth, M. W. Renner and K. M. Smith, J. Am. Chem. Soc., 1990, 112, 8851 Search PubMed; M. O. Senge, T. P. Forsyth, L. T. Nguyen and K. M. Smith, Angew. Chem., Int. Ed. Engl., 1994, 33, 2485 CrossRef CAS; K. M. Barkigia, J. Faler, M. D. Berber and K. M. Smith, Acta Crystallogr., Sect. C, 1995, 51, 511 CrossRef; D. J. Nurco, C. J. Medforth, T. P. Forsyth, M. M. Olmstead and K. M. Smith, J. Am. Chem. Soc., 1996, 118, 10918 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1: experimental procedures; 2: synthesis of 7–9 and 2; 3: Fig. S1; 4: Fig. S2; 5: references. See http://www.rsc.org/suppdata/cc/b2/b209789c/ |
| This journal is © The Royal Society of Chemistry 2003 |