A new entry to N-heterocyclic carbene chemistry: synthesis and characterisation of a triscarbene complex of thallium(I)†
Hidetaka
Nakai
,
Yongjun
Tang
,
Peter
Gantzel
and
Karsten
Meyer
*
University of California, San Diego, Department of Chemistry and Biochemistry, La Jolla, CA 92093, USA. E-mail: kmeyer@ucsd.edu
First published on 26th November 2002
Abstract
The synthesis and characterisation of a thallium(I) triscarbene complex of the chelating, tripodal carbene ligand 1,3,5-{tris(3-tert-butylimidazol-2-ylideno)methyl}-2,4,6-trimethylbenzene is reported, in which the thallium ion is coordinated by three N-heterocyclic carbene donors in a distorted trigonal planar environment.
Three-coordinate transition metal complexes with tripodal ligands are known to provide powerful platforms for small molecule activation and functionalisation;1 however, only a few examples of transition metal complexes using tripodal carbene ligands are reported in the literature.2 The considerable potential of N-heterocyclic carbenes for application in homogenous catalysis3 and small molecule activation, has triggered our efforts to develop a new class of metal complexes supported by tripodal N-heterocyclic carbene chelators. In this context, we are currently investigating the coordination chemistry of tripodal carbene-based ligands with an arene or a single atom as an anchoring unit. In 1994, the stable tripodal carbene ligand, 1,3,5-{tris(3-tert-butylimidazol-2-ylideno)methyl}-2,4,6-trimethylbenzene, timtmbtBu (1), was synthesised by Dias and Jin.4 Despite the reported isolation of the free carbene, this potential ligand has not been structurally characterised and there are no examples of metal complexes of this tripodal carbene system. Herein we report the first complex with this tridentate carbene ligand, [(timtmbtBu)TlI]+ (2), together with the crystal structures of free carbene 1 and its protonated precursor derivative [(timtmbnBu)Cl](Cl)2 (1a, see ESI†).
Free carbene 1 was synthesised via deprotonation of the corresponding imidazolium salt with NaH/KOtBu.4 Crystals of 1 suitable for X-ray diffraction analysis were grown from a saturated toluene solution at −35 °C.‡ The solid-state molecular structure of 1 is depicted in Fig. 1.
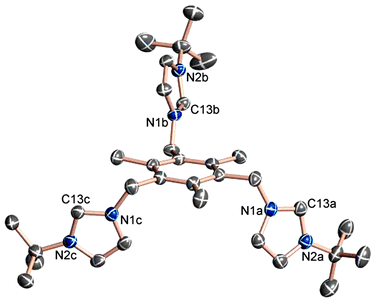 | ||
| Fig. 1 Solid-state molecular structure of free carbene 1. Hydrogen atoms are omitted for clarity, thermal ellipsoids at 40% probability. Selected bond lengths (Å) and angles (°): C13a–N1a 1.368(2), C13a–N2a 1.372(2), C13b–N1b 1.370(2), C13b–N2b 1.369(2), C13c–N1c 1.369(2), C13c–N2c 1.363(2); N1a–C13a–N2a 102.04(15), N1b–C13b–N2b 101.97(14), N1c–C13c–N2c 102.25(14). | ||
The three imidazole rings in 1 are arranged in an asymmetrical fashion with respect to the anchoring mesitylene ring. The electronic and molecular structures of the divalent carbon centres in 1 are similar to structures reported for other simple 1,3-disubstituted imidazole-2-ylidenes.5 The average C13–N distance at the carbene centre is 1.37 Å; the average N1–C13–N2 angle in the three imidazole entities is 102.0°. Our X-ray crystallographic results clearly show no evidence for any inter- or intramolecular interactions of the carbene centres of 1 in the solid-sate. In contrast, imidazolium salt 1a (see ESI† for crystal structure diagrams of 1a) crystallises in idealized C3 symmetry with all imidazolium moieties pointing up, thus providing a cavity for one of the three chloride anions.‡ The successful application of a wide variety of thallium complexes as ligand transfer reagents6 have stimulated our interest in preparing thallium complexes of carbene ligand 1 and its N2-substituted derivatives. Recently, Peters and coworkers have demonstrated a ligand transfer reaction of a Tl(I) complex supported by a tripodal phosphine ligand.7 Similarities of N-heterocyclic carbenes and organophosphine ligands in terms of their metal coordination chemistry are well known.3 These findings, together with our observations that the imidazolium precursors of this class of chelators form tripodal cavities and serve as anion hosts,8 have encouraged us to prepare a Tl(I) complex with the tridentate N-heterocyclic percarbene ligand 1.
The reaction of one equivalent of 1 with Tl(OTf) in THF at −35 °C led to the formation of the triscarbene thallium(I) complex, 2, in good yield (54%) as a white powder (Scheme 1).§
![Synthesis of [(timtmbtBu)TlI]+ (2) from free carbene (1).](/image/article/2003/CC/b209071f/b209071f-s1.gif) | ||
| Scheme 1 Synthesis of [(timtmbtBu)TlI]+ (2) from free carbene (1). | ||
The novel complex 2 is highly temperature sensitive. THF solutions of 2 can be kept at −35 °C for several days without decomposition. Allowing the reaction solutions to gradually warm to r.t., however, leads to black, unidentified decomposition products. This is in contrast to known Tl(III)–carbene complexes that have high thermal stability.9 Although the thermal instability and the reduced solubility of 2 at low temperatures impede thorough spectroscopic investigation, 2 was clearly identified in the low temperature 1H and 13C NMR spectra. The 1H NMR spectrum of free carbene 1 in THF-d8 at −35 °C exhibits resonances at δ 1.52, 2.47, 5.31, 6.54 and 7.08. When Tl(OTf) was added to a solution of free carbene 1 in THF-d8 at −35 °C, these signals clearly changed as follows: δ 1.47, 2.34, 5.33, 7.38 and 7.46. The most notable feature in the 1H NMR spectrum is the downfield shift of the imidazole CH resonances from δ 6.54 and 7.08 of free carbene 1 to δ 7.38 and 7.46 in spectra of samples of complex 2. However, we were unable to observe potential 203/205Tl–1H and/or 13C hyperfine features in our NMR spectra.9
The molecular structure of 2 was confirmed by X-ray crystallography. Colourless crystals 2(OTf)·2THF suitable for X-ray diffraction analysis were obtained by cooling a saturated THF solution at −35 °C overnight.‡Fig. 2 shows the solid-state molecular structure of cation 2.
 | ||
| Fig. 2 Two views of the solid-state molecular structure of 2 in crystals of 2(OTf)·2THF. Hydrogen atoms and co-crystallised solvent molecules are omitted for clarity, thermal ellipsoids at 40% probability. Selected bond lengths (Å) and angles (°): Tl1–C13a 2.979(4), Tl1–C13b 2.985(5), Tl1–C13c = 2.893(4), C13a–N1a 1.358(6), C13a–N2a 1.368(5), C13b–N1b 1.361(6), C13b–N2b 1.361(6), C13c–N1c 1.360(5), C13c–N2c 1.369(5); C13a–Tl1–C13b 124.05(13), C13a–Tl1–C13c 118.90(12), C13b–Tl1– C13c 109.65(13), N1a–C13a–N2a 102.6(4), N1b–C13b–N2b 102.9(4), N1c–C13c–N2c 102.5(3). | ||
The tripodal carbene ligand 1 coordinates the thallium cation in the expected tridentate conformation. The thallium(I) ion is displaced 0.466 Å out of a trigonal plane formed by the three carbenic centres and is positioned 2.919 Å above the arene anchoring unit. Comparison of the 13C resonances of the free carbene ligand with those of the thallium complex imply that there is no significant bonding interaction between the arene fragment and the Tl(I) ion. This observation is further supported by a DFT calculation (ESI†) carried out on the cationic thallium complex. Close examination of the frontier orbitals reveals no orbital interaction between the arene moiety and the metal ion. All three imidazole rings are almost perpendicular to the mesitylene entity, however, the solid-state molecular structure suggests that the three carbene centres in 2 are not symmetrically bound to the Tl(I) ion: the Tl1–C13c distance, 2.893(4) Å, is significantly shorter than the Tl1–C13a and Tl1–C13b distances (2.979(4) and 2.985(5) Å, respectively). The dihedral angles C(mesitylene)–C(methylene)–N1–C13 are 3.99, 4.91 and 32.86° for C13a, C13b and C13c, respectively. Despite the observed differences within the imidazole-2-ylidene units of 2, the divalent carbon centres are electronically identical (see bond lengths and angles in Fig. 2). A space-filling model helps to rationalize the deviation from an ideal C3 symmetry. It is obvious that this deviation is due to the large thallium(I) ion and the sterically demanding tert-butyl groups bound to N2. It therefore seems reasonable to conclude that the disturbed symmetry observed in the solid-state structure of 2 is due to steric or crystal packing effects rather than electronic origin. Accordingly, we are currently pursuing Tl(I) chemistry of other neutral tripodal carbene ligands conceptually but not sterically related to that described herein.
In summary, we have synthesised and characterised a triscarbene complex of TI(I). This is the first example of a Tl(I)-carbene complex as well as the only reported complex of this class of percarbene chelator. We are currently investigating the potential of this Tl(I) species as a ligand transfer reagent to low valent transition and actinide metal ions.
For financial support we gratefully acknowledge the University of California, San Diego.
Notes and references
- D. M. Jenkins, T. A. Betley and J. C. Peters, J. Am. Chem. Soc., 2002, 124, 11238 CrossRef CAS; L. H. Gade, Acc. Chem. Res., 2002, 35, 575 CrossRef CAS; R. R. Schrock, Acc. Chem. Res., 1997, 30, 9 CrossRef CAS.
- R. Fränkel, U. Kernbach, M. B. -Christianopoulou, U. Plaia, M. Suter, W. Ponikwar, H. Nöth, C. Moinet and W. P. Fehlhammer, J. Organomet. Chem., 2001, 617–618, 530 CrossRef CAS; U. Kernbach, M. Ramm, P. Luger and W. P. Fehlhammer, Angew. Chem., Int. Ed., 1996, 35, 310 CrossRef CAS.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed., 1997, 36, 2162 CrossRef CAS.
- H. V. R. Dias and W. Jin, Tetrahedron Lett., 1994, 35, 1365 CrossRef CAS.
- A. J. Arduengo, Acc. Chem. Res., 1999, 32, 913 CrossRef; A. J. Arduengo, H. Bock, H. Chen, M. Denk, D. A. Dixon, J. C. Green, W. A. Herrmann, N. L. Jones, M. Wagner and R. West, J. Am. Chem. Soc., 1994, 116, 6641 CrossRef; A. J. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530 CrossRef CAS.
- C. Janiak, J. Prakt. Chem., 1998, 340, 181 CAS; G. Parkin, Adv. Inorg. Chem., 1995, 42, 291 Search PubMed; C. Janiak, Main Group Met. Chem., 1998, 21, 33 Search PubMed.
- I. R. Shapiro, D. M. Jenkins, J. C. Thomas, M. W. Day and J. C. Peters, Chem. Commun., 2001, 2152 RSC.
- H. Ihm, S. Yun, H. G. Kim. J. K. Kim and K. S. Kim, Org. Lett., 2002, 4, 2897 CrossRef CAS; K. Sato, S. Arai and T. Yamagishi, Tetrahedron Lett., 1999, 40, 5219 CrossRef CAS.
- M. L. Cole, A. J. Davies and C. Jones, J. Chem. Soc., Dalton Trans., 2001, 2451 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: structure of [1a]2·5H2O, orbital interactions in 2, and computational details. See http://www.rsc.org/suppdata/cc/b2/b209071f/ |
| ‡ Crystallographic details for [1a]2·5H2O: C66H109.43Cl6N12O5, M = 1363.78, colourless plate, T = 101(2) K, monoclinic, space group P21/n, a = 19.0580(9), b = 17.9298(8), c = 23.6229(11) Å, β = 113.6460(10)°, V = 7394.4(6) Å3, Z = 4, R1 = 0.0653 [I > 2σ(I)], GOF = 0.949. CCDC 193368. Crystallographic details for 1: C33H49.25N6, M = 530.03, colourless hexagonal, T = 100(2) K, monoclinic, space group P21/n, a = 12.0758(7), b = 11.1310(6), c = 23.7831(14) Å, β = 100.5090(10)°, V = 3143.2(3) Å3, Z = 4, R1 = 0.0425 [I > 2σ(I)], GOF = 1.037. CCDC 193367. Crystallographic details for 2(OTf)·2THF: C42H64F3N6O5STl, M = 1026.42, colourless needle, T = 100(2) K, monoclinic, space group P21/c, a = 12.8495(12), b = 17.1020(16), c = 20.8654(19) Å, β = 97.580(2)°, V = 4545.1(7) Å3, Z = 4, R1 = 0.0405 [I > 2σ(I)], GOF = 1.005. CCDC 193366. See http://www.rsc.org/suppdata/cc/b2/b209071f/ for crystallographic data in CIF or other electronic format. |
| § Synthesis and spectroscopic data: for 2·OTf: a cooled (−35 °C) THF solution (5 cm3) of Tl(OTf) (177 mg, 0.5 mmol) was added to a cooled (−35 °C) solution of timtmbtBu (264 mg, 0.5 mmol) in THF (5 cm3). The mixture was stored for 12 h at −35 °C. The resulting white precipitate was filtered off, washed with cold THF, and dried in vacuum (yield: 237 mg, 54%). Recrystallisation of the crude product from a saturated solution of THF at −35 °C yielded needle-shaped crystals of 2(OTf)·2THF suitable for X-ray diffraction analysis. 1H NMR (500 MHz, THF-d8, −35 °C): δ 1.47 (s, C(CH3)3, 27H), 2.34 (s, Ar–CH3, 9H), 5.33 (s, CH2, 6H), 7.38 (br, NCH, 3H) and 7.46 (br, NCH, 3H). 13C NMR (500 MHz, THF-d8, 0 °C): δ 16.9 (Ar–CH3), 31.5 (CCH3), 49.6 (Ar–CH2), 57.3 (CMe3), 118.3 (NCC), 121.1 (CCN), 134.8 (Ar-C), 140.2 (Ar-C). For 1: 13C NMR (500 MHz, THF-d8, 0 °C): δ 16.9 (Ar–CH3), 31.5 (CCH3), 50.3 (Ar–CH2), 56.2 (CMe3), 116.1 (NCC), 117.5 (CCN), 134.1 (Ar-C), 139.0 (Ar-C), 215.2 (NCN). |
| This journal is © The Royal Society of Chemistry 2003 |