meso-Tetrakis[o-(N-methyl)pyridinium]porphyrin ensembles with axially coordinated cyclodextrin-penetrating phenethylimidazole: reversible dioxygen-binding in aqueous DMF solution
Teruyuki
Komatsu
a,
Shoichi
Hayakawa
b,
Eishun
Tsuchida
a and
Hiroyuki
Nishide
*b
aAdvanced Research Institute for Science & Engineering, Waseda University, Tokyo 169-8555, Japan
bDepartment of Applied Chemistry, Waseda University, Tokyo 169-8555, Japan. E-mail: nishide@waseda.jp; Fax: +81 3-3209-5522; Tel: +81 3-3200-2669
First published on 21st November 2002
Abstract
α-Cyclodextrin (αCD)-penetrating 2-methyl-1-phenethylimidazole coordinates to the zinc(II) and iron(II) complexes of meso-tetrakis[o-(N-methyl)pyridinium] porphyrinate, giving non-covalently linked αCD-porphyrin ensembles; the iron(II) complex can reversibly bind and release dioxygen in aqueous DMF solution.
Modified porphyrinatoiron(II) complexes with a highly-complicated structure, which are prepared by general organic synthetic procedures, namely covalent bonding, have been extensibly studied to mimic the diverse reactivities of hemoproteins.1,2 In particular, the designs of single-face or double-face encumbered models have been a topic of great interest for the preparation of dioxygen (O2)-carrying hemes as hemoglobin and myoglobin analogues.3,4 Based on these significant efforts, we now recognize that two crucial factors are necessary for the dioxygenation of the synthetic heme; (i) bulky-substituents on the porphyrin ring plane to prevent μ-oxo dimer formation, and (ii) a hydrophobic environment to exclude the protons, especially in aqueous media.5 However, a great deal of labor is generally required to introduce the encumbrance on the porphyrin macrocycle and the total synthetic yields are rather low. If a suitable molecular structure, which provides the O2-binding capability to the porphyrin platform, is constructed by non-covalent bond formations in water, a totally new class of porphyrin architectures will appear in this chemistry. We report herein for the first time the formation of meso-tetrakis[o-(N-methyl)pyridinium]porphyrinato-zinc(II) and -iron(II) ensembles with axially coordinated α-cyclodextrin-penetrating 2-methyl-1-phenethylimidazole (αCD-MPIm), and the reversible O2 binding to the iron(II) complex in aqueous DMF solution (Fig. 1). We have employed an αCD to prepare the water-soluble bulky proximal base, and its binding to the flat tetracationic porphyrinato-iron(II) leads to a stable O2-adduct formation. This is the first example of dioxygenation of a non-covalently linked supramolecular architecture of a cyclodextrin-heme complex.
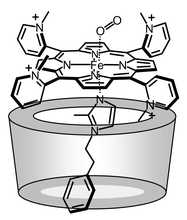 | ||
| Fig. 1 The possible dominant αααβ structure of the dioxygenated 1c(αCD-MPIm) ensemble. | ||
meso-Tetrakis(o-pyridyl)porphine (TPyP), prepared by Adler’s method,6 was reacted with CH3I in CHCl3–EtOH solution to yield the quaternarized 5,10,15,20-tetrakis[o-(N-methyl)pyridinium]porphine tetraiodide (47%). The Zn(II) insertion was carried out using Zn(OAc)2 in MeOH, affording the Zn(II) complex (1a). In the case of the iron complex (1b), the central metal was formerly introduced to TPyP by FeBr2 in DMF, and then quaternarized using CH3I. Both materials were finally passed through an ion exchange column (Dowex 1-2X) to convert their counter anions to Cl−. Analytical data for all the compounds were satisfactorily obtained.
As Miskelly et al. reported, four rotational atropisomers of 1a could be separated on silica gel using an eluent of 2-butanone–conc. aq. NH3–NH4PF6–N-methylimidazole (MIm).7 However, attempts to separate these isomers without PF6− counter anions and axially coordinated MIm were unsuccessful. Anyhow, it is true that the αααβ isomer is the most dominant species of 1a (ca. 50%) at thermal equilibrium.8
From the aqueous suspension of αCD and MPIm (20-fold molar excess), the equivalent inclusion compound, αCD-MPIm, was isolated as a white solid. The 1H NMR spectrum, where all signals were carefully assigned by 1H–1H COSY, showed that (i) it exactly consists of a 1∶1 complex of the host and guest molecules, and (ii) MPIm is incorporated into the apolar cavity of the αCD on the basis of the upfield shifts of the proton signals for the 3- and 5-positions inside the αCD ring; Δppm = +0.075 and +0.049 for 3H and 5H, respectively. However, the magnitudes of these shifts were relatively small compared to the previously reported examples.9 The molecular length of MPIm (9.5 Å) is longer than the depth of the αCD (6.7 Å), therefore, the ethylene moiety of MPIm may be located in the middle of the cyclic hexaglucopyranose. Hence, the ring current shift observed in the inner-H of αCD by the phenyl and imidazolyl groups is somewhat small. The FAB-MS also demonstrated a molecular-mass ion peak of αCD-MPIm at 1159.9 [M+]. The hydrophobic and dipole–dipole interactions are probably responsible for the driving force of this inclusion.10 The MOPAC calculations suggested that the conformer, in which the phenyl ring of MPIm (4.2 Debye) is oriented to the secondary hydroxyl side (the narrower rim) of αCD (9.4 Debye), is energetically more favorable than the reverse one.
The addition of this αCD-MPIm ligand to the aqueous solution of 1a gave a five-N-coordinate Zn(II) complex (λmax: 430, 559, 594 nm). Although anionic tetraarylporphyrins are known to form 1∶2 complexes with β-cyclodextrin, the cationic porphyrins have no interaction with any cyclodextrin family.11 The binding constant of αCD-MPIm (KB = 1.0 × 102 M−1 in water) to 1a was nearly the same as that of the 1,2-dimethylimidazole (DMIm) (KB = 0.9 × 102 M−1), indicating that the αCD-complexation produced no effect on the axial imidazole association constant. The most remarkable observation in the αCD-MPIm binding to the Zn(II) center is inducing the rotation of the C–C bond between the meso-C and the pyridinium 1-C. In the 1H NMR spectrum of 1a itself in D2O, the signals of pyrrole β-H and the pyridinium 6-H appeared as a singlet at 8.85 and 9.25 ppm, respectively. On the contrary, after the αCD-MPIm coordination, they both became doublets and the pyridium 3-H signals significantly shifted upfield (Δppm: −0.12 ppm). Since the DMIm binding to 1a did not induce such dramatic changes, we concluded that the bulky αCD-MPIm coordination influences the atropisomer equilibrium of 1a in statistical distribution.12 The chemical shifts for 2-CH3 hydrogens of the pyridinium groups in 1a(αCD-MPIm) might be more informative on which isomers exist,7 but they were interfered with by the αCD signals.
The ferric 1b was reduced to the corresponding ferrous complex (1c) in water by adding a two-fold molar excess of aqueous Na2S2O4 under an N2 atmosphere with αCD-MPIm. The UV-vis absorption spectrum showed the typical five-N-coordinate high-spin Fe(II) complex (λmax: 431, 533, 562 nm).3,4 However, upon exposure to O2 gas, 1c(αCD-MPIm) was oxidized to the ferric state even at low temperature (5 °C).
In a DMF–water (3/2, v/v) solution, the identical five-N-coordinate complex (λmax: 435, 538, 564, 621 nm) of 1c(αCD-MPIm) was formed under an N2 atmosphere as well, and the obtained complex was stable in the range of 10 μM–1 mM at 5–40 °C (Fig. 2). After bubbling O2 gas through this solution, the UV-vis absorption immediately changed to that of the O2-adduct complex [λmax: 422, 542, 570 (sh.) nm] at 5 °C.3–5 This dioxygenation was sufficiently kinetically stable, and reversibly observed depending on the O2 partial pressure. After the addition of CO, 1c(αCD-MPIm) produced a very stable carbonyl complex [λmax: 420, 535, 564 (sh.) nm]. The resulting O2 and CO adduct species are both diamagnetic and the 1H NMR spectra showed characteristics of S = 0.13 Oxidation to the Fe(III)porphyrin slowly took place; the final product was the Fe(III)OH complex with a λmax at 415 and 589 nm.14 It is quite remarkable that the oxidation process obeyed first-order kinetics (half-life was ca. 40 min at 5 °C) even under a relatively low O2-partial pressure (ca. 20 Torr) (Fig. 2 inset). The positively charged pyridinium groups at the porphyrin periphery could prevent μ-oxo dimer formation by an electrostatic repulsion. Neutral 5,10,15,20-tetraphenylporphyrinato-iron(II)(αCD-MPIm) [FeTPP(αCD-MPIm)] rapidly oxidized after the O2 bubbling under the same conditions. We considered that the αCD-MPIm coordination to the αααβ and αααα isomers of 1c took place from the β-side of the porphyrin plane by steric hindrance to the 2-CH3 groups of the pyridinium rings. As a result, there were at least two pyridinium cations surrounding the O2-coordination site of 1c(αCD-MPIm), and then the proton driven oxidation was retarded.
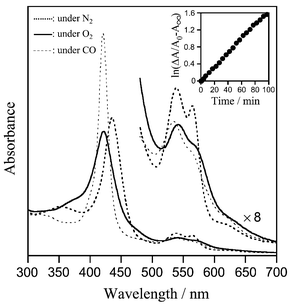 | ||
| Fig. 2 Visible absorption spectra of the 1c(αCD-MPIm) ensemble and its O2-, CO-adduct complexes in DMF–H2O (3/2 v/v) solution at 5 °C. The inset demonstrates the first-order plots of the absorption decay at 542 nm (O2-adduct species). | ||
The three-dimensional structure of the dioxygenated 1c(αCD-MPIm) ensemble was simulated by molecular dynamics calculations.15 The significant properties of the architecture are: (i) MPIm penetrates the cavity of αCD and half of the imidazole- and phenyl-rings are forced out from the cyclic hexaglucopyranose, which supports the 1H NMR spectral data, (ii) the meso-pyridinium groups and the rim of the αCD bucket contact within the van der Waals distance, and (iii) the imidazole coordination angle does not distort and is identical to that observed in the same calculation for other dioxygenated FeTPP derivatives.
In conclusion, the non-covalently linked αCD-porphyrin ensemble consisting of the simple flat tetracationic-porphyrinato-iron(II) and αCD-penetrating proximal imidazole showed the following unique characteristics. (i) The synthetic yield of the architecture based on the porphyrin is in principle ≈100%. (ii) The O2-adduct complex of 1c(αCD-MPIm) is the first example of a new class of synthetic O2-carrying hemoprotein models which is constructed by non-covalent bond formations. The molecular O2 can bind from the aqueous side to the flat porphyrinato-iron(II) and no oxidation occurs in polar environment, because protons cannot reach the dioxygen active site. (iii) The obtained ensemble was easily dissociated by the addition of methanol, and each building block was withdrawn by gel column chromatography. Further investigations to evaluate the O2-binding behavior of these porphyrin architectures are now underway.
This work was partially supported by a Grant-in-Aid for Scientific Research (No. 13650938, 13031072) and COE Program ‘Practical Nano-Chemistry’ from MEXT, Japan.
Notes and references
- For a review, see: for example, The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, Oxford, 1999, vol. 4 Search PubMed.
- (a) Y. Kuroda, T. Hiroshige, T. Sera, Y. Shiroiwa, H. Tanaka and H. Ogoshi, J. Am. Chem. Soc., 1989, 111, 1912 CrossRef CAS; (b) Y. Naruta, F. Tani and K. Maruyama, Chem. Commun., 1990, 1378 RSC; (c) H.-Y. Zhang, A. Blasko, J.-Q. Yu and T. C. Bruice, J. Am. Chem. Soc., 1992, 114, 6621 CrossRef CAS; (d) T. Sasaki and Y. Naruta, Chem. Lett., 1995, 663 CAS; (e) J. P. Collman, M. Rapta, M. Bröring, L. Raptova, R. Schwenninger, B. Boitrel, L. Fu and M. L’Her, J. Am. Chem. Soc., 1999, 121, 1387 CrossRef CAS.
- M. Momenteau and C. A. Reed, Chem. Rev., 1994, 94, 659 CrossRef CAS and references therein.
- (a) J. P. Collman and L. Fu, Acc. Chem. Res., 1999, 32, 455 CrossRef CAS; (b) E. Tsuchida, T. Komatsu, S. Kumamoto, K. Ando and H. Nishide, J. Chem. Soc., Perkin Trans. 2, 1995, 747 RSC; (c) T. Komatsu, K. Sano and E. Tsuchida, Chem. Commun., 1998, 977 RSC; (d) D. L. Jiang and T. Aida, Chem. Commun., 1996, 1523 RSC; (e) A. Kossanyi, F. Tani, N. Nakamura and Y. Naruta, Chem. Eur. J., 2001, 7, 2862 CrossRef CAS.
- (a) E. Tsuchida, T. Komatsu, K. Arai and H. Nishide, Chem. Commun., 1993, 730 RSC; (b) E. Tsuchida, T. Komatsu, K. Arai, K. Yamada, H. Nishide, C. Böttcher and J.-H. Fuhrhop, Langmuir, 1995, 11, 1877 CrossRef CAS; (c) T. Komatsu, M. Moritake, A. Nakagawa and E. Tsuchida, Chem. Eur. J., 2002, 8 CrossRef CAS , in press.
- J. B. Kim, A. D. Adler and F. R. Longo, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. 1, p. 85 Search PubMed.
- T. Kaufmann, B. Shamsai, R. S. Lu, R. Bau and G. M. Miskelly, Inorg. Chem., 1995, 34, 5073 CrossRef CAS.
- R. A. Freitag and D. G. Whitten, J. Phys. Chem., 1983, 87, 3918 CrossRef CAS.
- (a) L. Bender and M. Komiyama, in Bioinorganic Chemistry, ed. E. E. van Tameleon, Academic Press, New York, 1977, vol. I, ch. 2 Search PubMed; (b) ) Saenger, Angew. Chem., Int. Ed. Engl., 1980, 19, 344 CrossRef.
- M. Kitagawa, H. Hoshino, M. Sakurai, Y. Inoue and R. Chujo, Bull. Chem. Soc. Jpn., 1988, 61, 4225 CAS.
- K. Kono, N. Tanaka, H. Minamizono and Y. Kawakita, Chem. Lett., 1996, 925.
- R. Fiammengo, P. Timmerman, F. de Jong and D. N. Reinhoudt, Chem. Commun., 2000, 2313 RSC.
- J. P. Collman, J. I. Brauman, K. M. Doxsee, T. R. Halbert, E. Bunnenberg, R. E. Linder, G. N. LaMar, J. D. Gaudio, G. Lang and K. Spartalian, J. Am. Chem. Soc., 1980, 102, 4182 CrossRef CAS.
- D. Lexa, M. Momenteau, J.-M. Saveant and F. Xu, Inorg. Chem., 1985, 24, 122 CrossRef CAS.
- The esff forcefield simulation was performed using an Insight II system (Molecular Simulations Inc.). The structure was generated by alternative minimizations and annealing dynamic calculations from 298 to 100 K. Dielectric constant was fixed at 54.5 D, corresponding to DMF–H2O (3/2 v/v) solution.
| This journal is © The Royal Society of Chemistry 2003 |