A tripod ligand as new sensitiser for the enzyme amplified lanthanide luminescence determination of esterase
Tanja
Steinkamp
a,
Florian
Schweppe
b,
Bernt
Krebs
b and
Uwe
Karst
*a
aUniversity of Twente, Department of Chemical Analysis and MESA+ Research Institute, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: u.karst@ct.utwente.nl; Fax: ++31-53-489-4645
bWestfälische Wilhelms-Universität Münster, Institut für Anorganische und Analytische Chemie, Wilhelm-Klemm-Str. 8, 48149, Münster, Germany
First published on 27th November 2002
Abstract
Screening of a small library of tripod ligands resulted in the discovery of bis(2-pyridylmethyl)-(2-hydroxybenzyl)amine (HL1) as a new sensitiser, which is able to transfer its excitation energy to terbium(III). After synthesis of the acetic acid ester of HL1, a highly selective method for the determination of porcine liver esterase by means of enzyme amplified lanthanide luminescence (EALL) was developed. Enzyme-catalysed cleavage of the ester results in the formation of HL1. After excitation at 297 nm, the characteristic emission of Tb(III) at 545 nm is observed and used to determine the esterase concentration. In contrast to existing EALL methods, this method may be carried out at neutral pH and without further additives. Limit of detection for porcine liver esterase is 10−9 mol l−1 and limit of quantification is 3 × 10−9 mol l−1. A linear calibration range of two decades starting at the limit of quantification is observed.
Introduction
The two most promising approaches for reducing the limits of detection in bioassays are improvements of the biological components and of the detection scheme. For the latter, the long lived fluorescence of lanthanide chelates has gained increasing attention in recent years.1,2 The possibility for time resolved measurements combined with large Stokes’ shifts often exceeding 250 nm ensure the reduction of unspecific background signal to an absolute minimum.3 Enzyme amplified lanthanide luminescence (EALL) combines the advantages of lanthanide luminescence with a previous enzymatic reaction, thus leading to a significant amplification in signal intensity. So far, mainly salicylates have been employed for this technique. While 5-fluorosalicylic phosphate is not able to transfer excitation energy to Tb(III), its alkaline phosphatase-catalysed hydrolysis product 5-fluorosalicylate acts as a strong sensitiser. Alkaline phosphatase, xanthine oxidase, β-galactosidase and glucose oxidase may be analysed based on this and related approaches.4 A method for the determination of peroxidase has been described in literature as well.5The highest fluorescence intensities for lanthanide salicylate complexes are obtained at alkaline pH values,6 where the lanthanides easily tend to precipitate as their hydroxides. Therefore, EDTA is added as a co-complexing agent. As EDTA itself acts also as a very weak sensitiser and causes a small blank value, the extremely low limits of detection which could theoretically be achieved are not achieved in practice. Furthermore, the direct use of this detection method in biological matrices is complicated by the alkaline pH value. It is therefore desirable to develop an EALL-based system avoiding the above mentioned limitations.
Multidentate ligands are used for various applications in the area of inorganic chemistry.7–9 In this paper, we are the first to describe the use of tetradentate tripod ligands as sensitisers in EALL methods. As highly stable complexes are formed between these sensitising ligands and Tb(III), the addition of EDTA becomes unnecessary and background fluorescence is further decreased.
Experimental
Chemicals
All chemicals were purchased from Aldrich (Steinheim, Germany) in the highest purity available, except the following substances: bis-tris-propane (1,3-bis[tris(hydroxymethyl)methylamino)propane) and esterase from porcine liver (E.C. 3.1.1.1) were purchased from Sigma (Deisenhofen, Germany) in the highest purity available. Caesium chloride was obtained from AppliChem (Darmstadt, Germany) and triethylamine was from Fluka (Neu-Ulm, Germany). The ligands were synthesised according to literature.10Instrumentation
All quantitative determinations were carried out with a microplate reader from BMG LabTechnologies (Offenburg, Germany) with FLUOstar software version 2.10-0. The fluorescence spectra were recorded using a fluorescence spectrometer RF-5301 from Shimadzu (Duisburg, Germany) with RF 5301 PC software version 1.2.Synthesis of bis(2-pyridylmethyl)-(2-hydroxybenzyl)amine HL1
Synthesis of HL1 was carried out according to literature.10 A mixture of 4.39 g (21.8 mmol) bis(2-pyridylmethyl)amine, 4.98 g (21.8 mmol) 2-(bromomethyl)phenyl acetate and 4 ml triethylamine was dissolved in 90 ml of tetrahydrofurane and heated under reflux for 24 h. After cooling and filtration of the suspension, part of the filtrate was removed by vacuum evaporation. To the obtained red oil, 15 ml of ethanol and 5 ml of 40% sodium hydroxide solution were added. This mixture was subsequently refluxed for 12 h. To yield the free hydroxide, the mixture was neutralised (pH 8–10) with concentrated hydrochloric acid. Afterwards, it was extracted five times with 20 ml of diethyl ether. The organic phase was dried with magnesium sulfate, filtered off and the solvent was finally removed by means of vacuum evaporation. After column liquid chromatographic purification with a silica gel as stationary and ethyl acetate as mobile phase, the ligand was obtained as white powder. It was characterised by means of elemental analysis, 1H-NMR, 13C-NMR and IR.11Carboxylic acid ester synthesis of the ligand HL1
0.305 g (1 mmol) of HL1 was dissolved in 12 ml of acetonitrile, and 10 mmol of the respective carboxylic acid anhydride was added. After the addition of 279 μl (20 mmol) triethylamine and 2 mg of dimethylaminopyridine as catalyst, the mixture was refluxed for 72 h. After cooling down, the mixture was poured onto 50 ml of distilled water and extracted three times with dichloromethane. The organic phase was subsequently dried with sodium sulfate. Finally, the solvent was removed by means of vacuum evaporation. The obtained products were characterised by means of 1H-NMR, 13C-NMR, IR and MS (EI, 70 eV).Solution preparation
The esterase (from porcine liver E.C. 3.1.1.1) used as well as the various ligands and the esters of HL1 were dissolved in aqueous bis-tris-propane buffer (0.01 mol l−1) adjusted to a pH of 6.8. To ensure sufficient solubility of the ligands and the ester, 20% (v/v) dimethyl sulfoxide was added. The terbium chloride solution (0.01 mol l−1) was made with doubly distilled water.Time resolved fluorescence measurements
50 μl of a 0.01 mol l−1 Tb(III) solution was pipetted in each well of a 96 well microplate. Afterwards, 50 μl of ligand solution with concentrations between 10−3 mol l−1 and 10−8 mol l−1 were added. The fluorescence intensity was measured with a delay time of 50 μs and an integration time of 1 ms. The excitation wavelength was 297 nm, and emission was detected at 545 nm.Esterase determination with time resolved fluorescence
100 μl of esterase solution ranging from 1 u ml−1 to 10−5 u ml−1 was pipetted in each well of a microplate. Afterwards, 50 μl of ester solution (1 mmol l−1) was added to each well. After incubation at room temperature for 30 min, 50 μl of a 0.01 mol l−1 Tb(III) solution was added. Subsequent fluorescence measurements were accomplished as in case of the determination of ligand HL1.Results and discussion
Initially, a library of substances comprising twelve hydroxyphenyl-functionalised tripod ligands (structures listed in Table 1) was screened with respect to the ability of the individual compounds to serve as a sensitiser for the four lanthanide cations which emit in the visible area of the electromagnetic spectrum, namely Dy(III), Eu(III), Sm(III) and Tb(III). Although some of the investigated ligands turned out to be sensitisers for Tb(III), comparable effects for Dy(III), Eu(III) and Sm(III) were not observed at all. An experiment in which the Tb(III) cations of the luminescent complexes had to compete with added Eu(III) cations showed that the fluorescence intensity changed depending on the Eu(III) concentration. For a Tb3+ concentration of 0.01 mol l−1 for example, the fluorescence intensity is highest at an Eu3+ concentration of 0.001 mol l−1, is halved at an Eu3+ concentration of 0.01 mol l−1 and drops to a tenth of the original value at an Eu3+ concentration of 0.1 mol l−1. This strongly indicates that the other lanthanides also form complexes with the ligands, but that the ions’ acceptor levels are not compatible with the donor levels of the ligands.| Skeletal ligand structure:
|
|||||
|---|---|---|---|---|---|
| Ligand | R1 | R2 | R3 | Excitation maximum λex/nm | Relative fluorescence intensity at λex |
| HL1 | 2-Hydroxyphenyl | 2-Pyridyl | 2-Pyridyl | 297 | 100 |
| HL2 | 2-Hydroxy-3-tert-butylphenyl | 2-Pyridyl | 2-Pyridyl | 299 | 5 |
| HL3 | 2-Hydroxy-5-nitrophenyl | 2-Pyridyl | 2-Pyridyl | — | 0 |
| HL4 | 2-Hydroxyphenyl | 3-Methyl-2-pyridyl | 2-Pyridyl | 298 | 4 |
| HL5 | 2-Hydroxy-3-tert-butylphenyl | 3-Methyl-2-pyridyl | 2-Pyridyl | — | 0 |
| HL6 | 2-Hydroxy-5-nitrophenyl | 3-Methyl-2-pyridyl | 2-Pyridyl | — | 0 |
| H2L7 | 2-Hydroxyphenyl | 2-Pyridyl | 2-Hydroxyphenyl | 295 | 29 |
| H2L8 | 3-Dibromo-6-hydroxyphenyl | 2-Pyridyl | 2-Hydroxyphenyl | 301 | 14 |
| H2L9 | 3-Dibromo-6-hydroxyphenyl | 2-Pyridyl | 2-Hydroxy-5-nitrophenyl | — | 0 |
| H2L10 | 2-Hydroxy-5-nitrophenyl | 2-Pyridyl | 2-Hydroxy-5-nitrophenyl | — | 0 |
| H2L11 | 3,5-Dibromo-6-hydroxyphenyl | 2-Pyridyl | 2-Hydroxy-5-nitrophenyl | — | 0 |
| H2L12 | 2-Hydroxy-3-tert-butylphenyl | 2-Pyridyl | 2-Hydroxy-3-tert-butylphenyl | 339 | 2 |
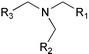
Considering the fluorescence intensity of the respective Tb(III) complexes, the ligand bis(2-pyridylmethyl)-(2-hydroxybenzyl)amine (HL1) is superior by a factor of three even to those ligands possessing similar structures. It is known from literature that relations between structural changes in the ligand’s backbone and the ability to serve as a lanthanide sensitiser are hard to predict.12 Regarding the tested ligands, it can generally be stated that complexes with nitro groups do not fluoresce at all. This can be associated with the significant change of donor and acceptor energy levels of the unsubstituted in comparison to the substituted ligands. As expected, sterically challenging alkyl moieties near the donor atoms decrease the fluorescence intensity.
Owing to the favourable spectroscopic properties, all further optimisation steps were accomplished using the Tb(III) complex of the ligand HL1. Excitation and emission spectra of this complex are shown in Fig. 1. Obviously, the excitation maximum of the complex is 297 nm. The emission spectrum is that of the terbium cation showing three narrowband maxima, the most intensive of which is located at a wavelength of 545 nm.
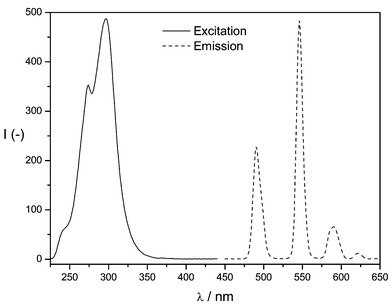 | ||
| Fig. 1 Excitation spectrum and emission spectrum of the Tb(III)–HL1 complex. | ||
Since the pH value has significant influence on the fluorescence intensity of the lanthanide complexes, it was varied in the following in the range between pH 6.6 and pH 7.6 in steps of 0.2 pH units (see Fig. 2). The optimum pH value for the buffer solution in which the HL1 lanthanide complex was dissolved was found to be pH 6.8, thus being advantageous especially for the application in biological matrices. Although YCl3 has been described to enhance luminescence intensity of various Tb(III) complexes,13 the addition of the latter had no influence on the fluorescence intensity of the given system. Moreover, an enhancement of intersystem crossing (ISC) does not occur for this system after addition of CsCl (heavy atom effect).14 The optimisation of fluorescence detection parameters resulted in an applied delay time of 50 μs and an integration time of 1000 μs.
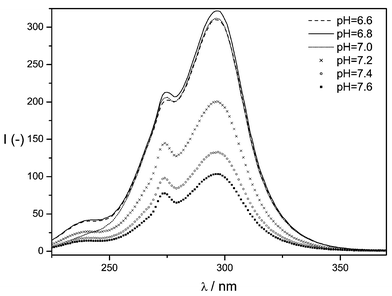 | ||
| Fig. 2 Influence of the pH value on the fluorescence intensity of the Tb(III)–HL1 system. | ||
For the development of a model detection system for hydrolytic enzymes, the acetic acid ester and the hexanoic acid ester of the ligand bis(2-pyridylmethyl)-(2-hydroxybenzyl)amine were synthesised. Fluorescence spectroscopic analysis of the esters, which was carried out by using the esters instead of HL1 in the optimised system described for HL1, revealed that both do not sensitise Tb(III), which is of particular advantage for the development of assays with extremely low background fluorescence.
Fig. 3 depicts the enzyme-catalysed hydrolysis of the acetic acid ester of HL1. The ester cleavage, in which the sensitising ligand is formed out of the non-sensitising ester through esterase-catalysed hydrolysis, as well as the subsequent fluorescence measurements are performed at a pH of 6.8. Thus, it is not necessary to change buffers during the complete analytical process. All further analytical parameters are listed in the Experimental section. With the procedure described there, a detection limit for esterase from porcine liver of 1 nmol l−1 is obtained. The limit of quantification is 3 nmol l−1, and the calibration range is linear over two decades starting from the limit of quantification. Repeated measurements (n = 8) of the same concentration resulted in relative standard deviations of 2.4%, 5.9% and 7.6% for concentrations of 10−7 mol l−1, 10−8 mol l−1 and 3 × 10−9 mol l−1, respectively. Using the more lipophilic hexanoic acid ester, a detection limit of 1 μmol l−1 is obtained. Therefore, the acetic acid ester is superior to the hexanoic acid ester by three decades of concentration. Due to the time resolved fluorescence measurements, the large Stokes shift and the narrow bands of metal ion fluorescence a considerable gain in selectivity is achieved, which provides the possibility of enzymatic analyses in complex biological matrices.
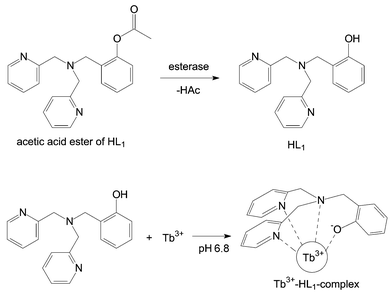 | ||
| Fig. 3 Reaction scheme for the esterase-catalysed hydrolysis of the acetic acid ester of ligand HL1with subsequent complex formation. | ||
Based on these promising results, a larger number of hydroxy-functionalised multidentate ligands will be screened in the future. Thus, it is foreseen that a sensitiser with even better limits of detection will be found. The current method shall be applied to activity screening of different hydrolytic enzymes using microplate technology and post-column detection after liquid chromatographic separation.
References
- E. F. Gudgin Dickson, A. Pollak and E. P. Diamandis, Pharmacol. Ther., 1994, 66, 207–235 CrossRef CAS.
- I. A. Hemmilä, Application of Fluorescence in Immunoassays, John Wiley and Sons, New York, 1991 Search PubMed.
- E. S. Lianidou, P. C. Ioannou and E. Sacharidou, Anal. Chim. Acta, 1994, 290, 159–165 CrossRef CAS.
- R. A. Evangelista, A. Pollak and E. F. Gudgin Templeton, Anal. Biochem., 1991, 197, 213–224 CrossRef CAS.
- J. Meyer and U. Karst, Analyst, 2001, 126, 175–178 RSC.
- T. K. Christopoulos and E. P. Diamandis, Anal. Chem., 1992, 64, 342–346 CrossRef CAS.
- R. Than, A. A. Feldmann and B. Krebs, Coord. Chem. Rev., 1999, 182, 211–241 CrossRef.
- M. Pascaly, M. Duda, F. Schweppe, K. Zurlinden, F. K. Müller and B. Krebs, J. Chem. Soc., 2001, 6, 828–837 Search PubMed.
- L. F. Szczepura, L. M. Witham and K. J. Takeuchi, Coord. Chem. Rev., 1998, 174, 5–32 CrossRef CAS.
- S. Ito, S. Nishino, H. Itoh, S. Ohba and Y. Nishida, Polyhedron, 1998, 17, 1637–1642 CrossRef CAS.
- F. Schweppe, PhD Thesis, University of Münster, Münster, Germany, 2000.
- E. P. Diamandis, Analyst, 1992, 117, 1879–1884 RSC.
- A. L. Jenkins and G. M. Murray, Anal. Chem., 1996, 68, 2974–2980 CrossRef CAS.
- R. Zhu and W. T. Kok, Anal. Chem., 1997, 69, 4010–4016 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2003 |