Membrane introduction mass spectrometry for monitoring complexation equilibria of β-cyclodextrin with substituted benzenes
Rodinei
Augusti
ab,
Maciej
Turowski
a and
R. Graham
Cooks
*a
aDepartment of Chemistry, Purdue University, West Lafayette, IN 47907-1393, USA. E-mail: cooks@purdue.edu; Fax: 1-765-494-0239; Tel: 1-765-494-5262
bDepartment of Chemistry, Federal University of Minas Gerais, Belo Horizonte/MG 31270-901, Brazil. E-mail: augusti@dedalus.lcc.ufmg.br; Fax: 55-31-3499-5700; Tel: 55-31-3499-5725
First published on 4th December 2002
Abstract
Membrane introduction mass spectrometry (MIMS) was used to monitor complexation reactions between β-cyclodextrin (CD) and a series of benzene derivatives in aqueous solution. The equilibrium constants for benzene, chlorobenzene, bromobenzene, iodobenzene, toluene, cyanobenzene and nitrobenzene were determined. The suitability of MIMS for monitoring complexation reactions of organic compounds with host molecules was demonstrated. Structure–activity relationship analysis shows that the inclusion phenomena are driven by a variety of chemical forces, of which hydrophobicity is predominant for non-polar compounds, but not the only factor for more polar ones.
Introduction
The molecular properties of cyclodextrins and their complexation properties make them of wide interest.1 These conically-shaped cyclic oligosacharides are characterized both by hydrophobic (inner cavity) and by hydrophilic (outer surface) properties. Gas-phase inclusion reactions with CD host molecules have been studied by mass spectrometry, e.g. by FT-ICR-MS-MS, and recently they have been thoroughly reviewed by Lebrilla2 and Schalley.3 Inclusion/complexation reactions with cyclodextrins in the liquid phase have been investigated using various analytical techniques including UV absorption,4 fluorescence lifetime,5 fluorimetry,6 NMR,7,8 XRD9 and HPLC.10 Use of these techniques is restricted in application to certain compounds (e.g. detectability of UV-VIS spectrometry excludes alkanes), and may be limited by methodology (e.g. X-ray spectroscopy cannot be used for kinetics measurements). Studies on the thermodynamics and kinetics of CD inclusion reactions have not previously employed membrane introduction mass spectrometry (MIMS) and the objective of this paper is to explore the advantages of this solution sampling/chemical analysis method.MIMS11 is applicable to real-time, in situ monitoring—especially of smaller, hydrophobic compounds—typically in aqueous medium.12,13 The basis of the MIMS method lies in the fact that volatile and semi-volatile organic compounds can permeate hydrophobic membranes, usually polydimethylsiloxanes (PDMS), while water, polar, and ionized species do so much more slowly if at all. The sample is delivered to one side of the membrane and analytes permeate the membrane arriving at the inner surface which is exposed to an evacuated mass spectrometry system. An inert gas flow (usually He) helps to desorb the analytes and transport them into the mass analyser. Analyte molecules are thus relatively easily separated from the aqueous matrix and there is no need for any other sample pre-treatment. Quantitative data are provided rapidly and, in favorable instances, structural information about unknown reaction products can be inferred. For example, studies have been reported on the chlorination by hypochlorite of phenol, di- and trihydroxybenzenes, nitrobenzene, toluene and other benzene derivatives14 in which chloroform formation was monitored by MIMS. The same technique was used to monitor the oxidation of benzene derivatives by Fenton’s reagent.15 Shang and Blatchley reported environmental application of MIMS in monitoring free chlorine and inorganic chloramines in water.16
In this study MIMS is used to determine the equilibrium constants for complexation reactions with β-cyclodextrin (CD). Use of MIMS might provide an additional tool for investigations in this field. Certainly the high sensitivity of MIMS is noteworthy: it provides detection of analytes in water down to trace levels (parts per million into the parts per quadrillion range).17 There is an expectation that this analytical methodology may allow investigation of complexation with CD by compounds where this is difficult or impossible to detect using other techniques. In addition, membrane introduction systems are relatively simply constructed and many geometries exist.18–21 They are easily mounted in many existing mass spectrometers (especially GC-MS instruments), after only minor instrument modification.22–24 For example, a membrane inlet system coupled with a low-cost quadrupole mass spectrometer was described for use in fermentation process monitoring.25
The current MIMS experiment relies on the fact that neither β-cyclodextrin, nor the CD-substrate complex, can permeate the PDMS membrane, only free substrate in solution does. The initial concentrations of cyclodextrin, [CD]i, and substrate (defined as X), [X]i are known and the equilibrium concentration of the free substrate, [X]eq is monitored by the MIMS experiment. The cyclodextrin-substrate complex [X·CD] can be determined from mass balance:
| [X·CD] + [X]eq = [X]i | (1) |
| K = [X·CD]/[X]eq[CD] = ([X]i − [X]eq)/[X]eq[CD] | (2) |
| CD + X → X·CD |
| K = (Ai – Aeq)/Aeq[CD] | (3) |
We have chosen to examine a series of benzene derivatives, as there is a lack of experimental data on inclusion equilibria with β-CD for this class of compounds.26 In addition, examination of an array of substrates could produce a set of data that might serve for structure–activity relationship (SAR) analysis. This would allow an investigation of the physicochemical factors responsible for inclusion (or lack of inclusion) into the cyclodextrin cavity. Numerous papers deal with the factors responsible for complexation with CD’s.26–30 A comprehensive review has appeared recently,26 but an overall model of inclusion has not yet been proposed. One of the most important driving forces has been assumed to be the hydrophobic interaction;31 however, other factors such as hydrogen bonding may also contribute.29 These questions are taken up in this study.
Experimental
Experiments were performed using a flow-injection MIMS system.32 Substrates were dissolved in purified water (50 kΩ cm; Barnstead Mega-Pure® system, Dubuque, Iowa, USA) in order to obtain 1–8.7 ppm solutions, which were then divided into two 10 mL volumetric flasks. The concentrations of the different analytes were chosen to be as low as possible and still provide adequate signals. To one of the two solutions ca. 200 mg of solid β-cyclodextrin was added and dissolved. The other served as a reference. The molar ratio of CD to the substrate was larger than 150∶1. Both flasks were sealed and kept at constant temperature for 24 h without shaking. The solutions were sampled using the MIMS system via an automated VALCO (Houston, Texas, USA) 10-port valve system, containing a 1 mL sampling loop. The MIMS system33 consisted of a 3 cm long tubular polydimethylsiloxane (PDMS) membrane (Dow Corning, Midland, MI) thermostated at 50 °C. The membrane was encapsulated in ¼ inch PTFE tubing (Swagelok™ standard), which was evacuated and interfaced to the mass spectrometer via a 10 cm PTFE capillary tube. Helium was used as the carrier gas. The solutions were introduced by peristaltic pump at 1 mL min−1 flow rate. The mass spectrometer used was a Finnigan-MAT ITS-40 ion trap, modified as described elsewhere.34Results and discussion
The temporal stability of the instrumental system was tested by pumping water through the MIMS system, followed by a nitrobenzene solution (0.5 ppm) which was passed continuously over a period of several hours. One can note the relatively constant signal level observed (Fig. 1). Fig. 2 reproduces the flow-injection profiles observed for injection of (a) toluene and (b) cyanobenzene solutions. Injections of the standard solution (S) were followed by injections of the reaction solution (R). Data such as this were taken over the course of several hours. The rise and fall times were different for various analytes, as expected. Data for the more slowly responding compounds were repeated on different days.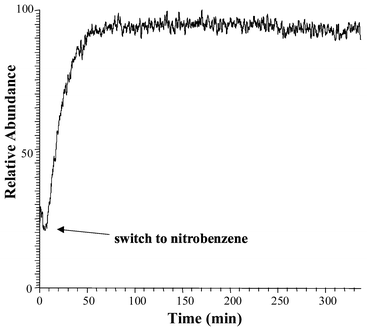 | ||
| Fig. 1 Time profile for continuous monitoring of the nitrobenzene ion signal (0.5 ppm concentration) demonstrating signal stability over a relatively long period of time. | ||
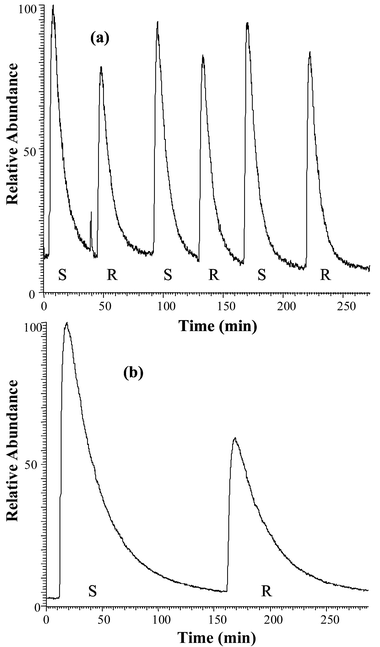 | ||
| Fig. 2 Time profiles (flow-injection analysis ‘chromatograms’) of (a) toluene and (b) cyanobenzene standard solutions (S) vs. reaction mixtures (R) with β-cyclodextrin. Each peak represents the total ion current response after 1 min of exposure to the membrane. Note the significant difference in rise and fall times for both analytes. | ||
Equilibrium constants, calculated according to eqn. (3), are presented in Table 1. The logK values for benzene were measured at concentrations of 0.5 and 5 ppm to establish that the analyte concentration did not influence the equilibrium values significantly. This was the case with the very high excess of CD used in these experiments. The confidence intervals for the equilibrium constants were calculated at the 95% confidence level using standard estimated errors for each analyte. Intuitively, the more hydrophobic the substrate is, the larger the inclusion reaction equilibrium constant expected, assuming a simple hydrophobic model. Previously however, this assumption could not be tested as no experimental data existed for the interactions of halogenated benzene derivatives with β-CD. Our findings are roughly in agreement with this expectation.
| Substrate | Concentration (ppm) | logK | CI (95%) | logP35 |
|---|---|---|---|---|
| Iodobenzene | 4.0 | 1.874 | 0.149 | 3.25 |
| Cyanobenzene | 7.8 | 1.661 | 0.004 | 1.56 |
| Bromobenzene | 2.0 | 1.643 | 0.303 | 2.99 |
| Chlorobenzene | 2.0 | 1.586 | 0.166 | 2.84 |
| Benzene | 5.0 | 1.529 | 0.082 | 2.13 |
| Benzene | 0.5 | 1.522 | 0.114 | 2.13 |
| Nitrobenzene | 7.8 | 1.421 | 0.149 | 1.85 |
| Toluene | 1.0 | 1.155 | 0.151 | 2.73 |
One should note the relatively large confidence intervals, attributed in part to experimental imperfections arising from headspace and membrane effects. It was noted during the course of many experiments that the standard solution signal had a slight tendency to decrease over time, especially for the more volatile analytes. The signal of the reaction mixture, however, remained constant, which is an excellent example of the protective properties of cyclodextrins, i.e. inclusion prevents loss of guest molecules by evaporation. As this is the first report on the use of MIMS for cyclodextrin inclusion reaction monitoring, we do not describe an extensive series of experiments, but rather delineate the capabilities of the methodology. Hydrophobic properties of halogenated benzenes, expressed in terms of logP, increase in parallel with the equilibrium rate of complexation with β-CD. Different behavior is noted, however, for toluene and two more polar benzene derivatives: nitrobenzene and cyanobenzene. The value of logP for toluene is slightly smaller than that for chlorobenzene and larger than that for benzene, yet the logK value for toluene is the smallest among all analytes studied. This is an unexpected phenomenon that will require more attention in the future.
Nitrobenzene and cyanobenzene are less hydrophobic than the other analytes, but they have relatively high logK values, especially cyanobenzene with an equilibrium constant close to that of iodobenzene. Previous findings showed that in the case of substrates possessing mixed hydrophobic/hydrophilic properties, the hydrophobic group inserts into the cyclodextrin cavity while the hydrophilic part remains outside, exposed to water.26 Exceptions to this are known for hydroxylated aromatic compounds (e.g. tyramine), where the substrate OH group penetrates deeply into the CD cavity and forms a hydrogen bond with a CD peripheral hydroxyl group, stabilizing the complex.29 In fact, comparison of literature data26 for phenols related to the substrates studied here indicates that their logK values are higher (as measured for β-CD in pure water), e.g. 3.4 for phenol, 2.40 for 4-chlorophenol, 2.98 for 4-iodophenol and 2.28, 2.54 or 3.0 (depending on the method) for 4-nitrophenol. A relatively high logK value for the relatively hydrophilic nitrobenzene and the very high logK and hydrophobicity values for cyanobenzene suggest that there may be a somewhat similar complex stabilization process involved in these cases. Again, however, further study is needed to confirm whether or not it involves hydrogen bond formation. Another factor that may contribute to complex stability is substrate size. Three different types of natural cyclodextrins (α, β and γ) with different cavity diameters (5.3 Å, 6.5 Å and 8.3 Å, respectively)26 show differences in the corresponding inclusion equilibrium constants. Among the compounds analysed in this work, only benzene can be compared with previously reported values. Values of logK for the 1∶1 benzene/CD complexes (in H2O at room temperature)26 are 1.50 for α-CD; 2.03 and 2.23 for β-CD (depending on the method) and 0.96 for γ-CD. This example demonstrates that there must be a certain ‘steric fit’ between the cyclodextrin cavity and substrate size in order to stabilize the inclusion complex.
There are additional factors that are indirectly related to the ‘size fit’ between substrate and CD: one involves H2O molecules residing inside the CD cavity before the inclusion reaction takes place, and the second, is associated with conformational changes of the cyclodextrin molecule itself upon complexation. It is estimated that β-CD usually accommodates 6 to 7 water molecules inside the cavity,36 but the number of molecules released upon complexation has not been calculated. Also the conformational changes of CD have not yet been quantitatively described. These two factors can obviously influence complex stability.
Conclusions
MIMS methodology is successfully implemented in monitoring cyclodextrin complexation reactions in the aqueous phase. The high MIMS sensitivity provides the considerable advantage of allowing investigation of substrates that are poorly soluble in water. Appropriate experimental arrangements may in future prove the usefulness of this technique for related kinetics and other thermodynamic measurements. The purpose of this work was to demonstrate the applicability of the MIMS methodology to the extensive research area of host–guest complexation reactions using cyclodextrin as exemplary host. As expected, the inclusion reaction equilibria correlate with hydrophobic properties for the series of non-polar halogenated benzenes series. The logK values recorded for more polar analytes suggest that the proposed complexation models should consider structural and conformational changes associated with the inclusion process, as well hydrogen-bonding capabilities with peripheral hydroxyl groups on the CD.Acknowledgements
This work was supported by the Indiana 21st Century Fund and by the Integrated Detection of Hazardous Materials (IDHM) Program. Support from the Brazilian National Research Council (CNPq) to R. Augusti is also acknowledged. We thank Dr Robert Noll for valuable discussions.References
- J. Szejtli, Chem. Rev., 1998, 98, 1743 CrossRef CAS.
- C. B. Lebrilla, Acc. Chem. Res., 2001, 34, 653 CrossRef CAS.
- C. A. Schalley, Mass Spectrom. Rev., 2001, 20, 253 CrossRef CAS.
- L. Caron, S. Tilloy, E. Monflier, J. M. Wieruszeski, G. Lippens, D. Landy, S. Fourmentin and G. Surpateanu, J. Inclusion Phenom. Macrocyclic Chem., 2000, 38, 361 CrossRef CAS.
- G. Nelson, G. Patonay and I. M. Warner, Anal. Chem., 1988, 60, 274 CrossRef CAS.
- Y. L. Loukas, V. Vraka and G. Gregoriadis, J. Pharm. Pharmacol., 1997, 49, 127 Search PubMed.
- M. Komiyama and H. Hirai, Bull. Chem. Soc. Jpn., 1981, 54, 828 CAS.
- G. Fronza, A. Mele, E. Redenti and P. Ventura, J. Org. Chem., 1996, 61, 909 CrossRef CAS.
- S. Z. Lin, N. Kohyama and H. Tsuruta, Ind. Health, 1996, 34, 143 Search PubMed.
- C. Ravelet, E. Peyrin, A. Villet, C. Grosset, A. Ravel and J. Alary, Chromatographia, 2001, 53, 624 Search PubMed.
- N. Srinivasan, R. C. Johnson, N. Kasthurikrishnan, P. Wong and R. G. Cooks, Anal. Chim. Acta, 1997, 350, 257 CrossRef CAS.
- R. C. Johnson, N. Srinivasan, R. G. Cooks and D. Schell, Rapid Commun. Mass Spectrom., 1997, 11, 363 CrossRef CAS.
- R. C. Johnson, R. G. Cooks, T. M. Allen, M. E. Cisper and P. H. Hemberger, Mass Spectrom. Rev., 2000, 19, 1 CrossRef CAS.
- R. V. R. A. Rios, L. L. Da Rocha, T. G. Vieira, R. M. Lago and R. Augusti, J. Mass Spectrom., 2000, 35, 618 CrossRef CAS.
- R. Augusti, A. O. Dias, L. L. Rocha and R. M. Lago, J. Phys. Chem. A, 1998, 102(52), 10723 CrossRef CAS.
- C. Shang and E. R. Blatchley III, Environ. Sci. Technol., 1999, 33, 2218 CrossRef CAS.
- S. Bauer, Trends Anal. Chem., 1995, 14, 202 CrossRef CAS.
- F. R. Lauritsen, Int. J. Mass Spectrom. Ion Processes, 1990, 95, 259 CrossRef CAS.
- S. Bohatka and H. Degn, Rapid Commun. Mass Spectrom., 1991, 5, 433 CAS.
- F. R. Lauritsen and R. A. Ketola, Anal. Chem., 1997, 69, 4917 CrossRef CAS.
- L. S. Riter, Z. Takats, L. Charles and R. G. Cooks, Rapid Commun. Mass Spectrom., 2001, 15(17), 1520 CrossRef CAS.
- S. Bauer, T. Griffin and J. Bauer, Int. J. Mass Spectrom. Ion Processes, 1996, 155, 107 CrossRef CAS.
- M. E. Bier, T. Kotiaho and R. G. Cooks, Anal. Chim. Acta, 1990, 231, 175 CrossRef CAS.
- P. Broz, E. Drbalkova, P. Janderka, P. Sitko and J. Vrestal, Chem. Listy, 2000, 94, 123 Search PubMed.
- S. Chauvatcharin, K. B. Konstantinov, K. Fujiyama, T. Seki and T. Yoshida, J. Ferment. Bioeng., 1995, 79(5), 465 Search PubMed.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875 CrossRef CAS and the references cited therein.
- L. Liu, K. S. Song, X. S. Li and Q. X. Guo, J. Inclusion Phenomena Macrocyclic Chem., 2001, 40(1–2), 35 Search PubMed.
- E. Junquera, D. Ruiz and E. Aicart, J. Colloid Interface Sci., 1999, 216, 154 CrossRef CAS.
- P. D. Ross and M. V. Rekharsky, Biophys. J., 1996, 71, 2144 Search PubMed.
- P. Irwin, J. Brouillette, A. Giampa1, K. Hicks, A. Gehring and S. Tu, Carbohydr. Res., 1999, 322, 67 CrossRef CAS.
- M. V. Rekharsky, M. P. Mayhew, R. N. Goldberg, P. D. Ross, Y. Yamashoji and Y. Inoue, J. Phys. Chem. B, 1997, 101, 87 CrossRef CAS.
- N. Kasthurikrishnan, R. G. Cooks and S. Bauer, Rapid Commun. Mass Spectrom., 1996, 10, 751 CrossRef CAS.
- M. A. LaPack, J. C. Tou and C. G. Enke, Anal. Chem., 1991, 63, 1631 CrossRef CAS.
- L. S. Riter, Z. Takats and R. G. Cooks, Analyst, 2001, 126, 1980 RSC.
- An on-line logP database is located at http://esc.syrres.com/interkow/physdemo.htm.
- K. Lindner and W. Saenger, Angew. Chem., Int. Ed. Engl., 1978, 17, 694 CrossRef.
| This journal is © The Royal Society of Chemistry 2003 |