Electrophoresis of DNA in ionic liquid coated capillary
Weidong
Qin
a and
Sam Fong Yau
Li
*ab
aDepartment of Chemistry, National University of Singapore, 119260, Republic of Singapore. E-mail: chmlifys@nus.edu.sg; Fax: +65-67791691
bCE Resources Pte Ltd, Block 51, Ayer Rajah Crescent, #06-08/09, Ayer Rajah Industrial Estate, Singapore, 139948, Republic of Singapore
First published on 5th December 2002
Abstract
An ionic liquid (IL) coated capillary was prepared and investigated for DNA separation. The electroosmotic flow of the capillary was reversed between pH 4.5 and 9. Below 900 base pairs the larger DNA fragment suffered more retardation in the IL coated capillary due to the increasing charge density of the fragment with size. In the presence of 4% hydroxyethylcellulose, the ΦX174 DNA-Hae III digest fragments were baseline separated in both IL- and polyacrylamide-coated capillary except for the fragments of 271 and 281 base pairs; while the analysis time was shorter in the IL-coated capillary. Our experiments indicated that the IL-coated capillary could work stably in the run buffer for at least 96 h with no notable deterioration in performance.
Introduction
Capillary electrophoresis (CE) is an increasingly-used tool for DNA separation; in fact it has been an attractive alternative to the conventional slab gel method owing to its high efficiency and quick separation.1–4 However, coating of the internal silica surface, either by covalent bonding or adsorption, is usually needed to suppress electroosmotic flow (EOF) and DNA–wall interaction5 so that both recoveries of the targets and reproducibility of migration times can be improved. Polymers such as linear polyacrylamide (PA) and its derivatives,4,6,7 cellulose and its derivatives,8–10 poly(ethylene oxide) (PEO)11–13 and polyvinylpyrrolidone14 have been used as sieving matrices and some of them were employed as self-coating (adsorption) reagents. Self-coating is compelling because of its simplicity. But for the capillary coated by static adsorption, its performance deteriorates with multiple runs and rinsing with the coating polymer between runs is usually required. The covalent coating exhibits longer lifetime than the dynamic coating and needs less maintenance.15 Since Hjerten proposed silanization of the capillary surface,16 a number of new materials or methods have been reported for the covalent bonding.17–33PEO and HEC, the weak self-coating polymers, are often used sieving matrices for DNA separation and are efficient for large-size fragments. However, the large DNA fragments migrate at very low speeds due to the sieving effect of the matrix and their migration times are usually very long in the presence of the large opposite residual EOF; sometimes the fragments cannot even reach the detector. In order to improve the separation speed, the silica surface is usually covalently coated to reduce the EOF. But as we observed in our experiment and reported by other authors, there is still residual EOF after the coating procedure. Another approach for fast analysis is separating DNA fragments in presence of EOF13,34,35 in which the analytes are drawn to the detector by the high-velocity EOF. Detection of the large-fragment DNA will not be a problem because they are drawn out first, but the migration times for the small fragments will be long. Moreover, due to the low concentration of sieving matrix used in order for the high residual EOF, the separation was only fast and better for the large DNA fragments under normal mode.34
For the capillary coated with charged materials, the coating may act as both stationary phase and ion exchanger. The (small) oppositely charged species are separated under a different mode from conventional capillary zone electrophoresis (CZE), and was termed as ion-exchange open tubular capillary electrochromatography (IE-OTCEC). The technique was studied theoretically and experimentally in the past couple of years.36–39 Generally, cationic coating is not chosen for DNA separation because the negatively charged DNA fragments will strongly interact with or even adsorb onto the wall. But adsorption of macromolecules onto the oppositely charged wall does not occur under all circumstances. Protein, which is more prone to suffer wall-adsorption than DNA, will not be adsorbed onto a surface if the association constant of the protein–surface interaction is less than 103 dm3 mol−1.40 If the silica surface is covered with cationic coating, the EOF will be reversed so that it moves codirectionally with the DNA fragments. There may exists a chance that the analysis times of the fragments are shortened provided that the electrostatic interaction between DNA fragments and the cationic coating is efficiently controlled. Such coating may be an alternative approach for fast separation of these multivalent macromolecules.
Dialkylimidazolium based ionic liquids (ILs) have generated increasing interests in the past decades, studies have been carried out on their feasibilities as low-cost electrolytes in solar battery, non-flammable solvents for synthesis, stationary phases in gas chromatography, and electrolytes in capillary electrophoresis as well.41–46 In the work of Valkenberg et al.,47 one IL was coated onto the silica powder surface for catalysis study. Although the IL cation also contains an imidazole ring, it remains cationic in alkaline buffer for DNA separation; moreover it is transparent at the DNA detection wavelength.
We report in this work the preparation of a IL-coated capillary (ILCC) and assessment of its properties relating to DNA separation. Our experiments indicated that the interaction between DNA and the IL-coating was fragment-size dependent. In the presence of hydroxyethylcellulose (HEC), DNA separation could be accomplished with shorter analysis time in ILCC than in PA-coated capillary (PACC).
Materials and methods
Chemicals
The ΦX174 DNA-Hae III digest was purchased from Sigma (Saint Louis, MO, USA). Acrylamide, γ-methacryloxypropyltrimethoxysilane, N,N,N′,N′-tetramethylenediamine, 3-chloropropyl-trichlorosilane (CPTCS), HEC (CAS number: 9004-62-0), imidazole and potassium persulfate were obtained from Fluka (Buchs, Switzerland). Dimethyl sulfoxide (DMSO) was supplied by Merck (Darmsradt, Germany). 1-Chlorodecane was product of Aldrich (Milwaukee, WI, USA). Polyvinylpyrrolidone (PVP, average molecular mass: 1000000) was product of Polysciences (Warrington, PA, USA). Tris–boric acid–EDTA (TBE) solution containing 445 mM Tris, 445 mM boric acid and 10 mM EDTA at pH 8.3 was prepared and diluted to the desired concentrations for use. Stock solutions of PVP and HEC of 10% (w/v) each were prepared by adding weighted PVP or HEC to distilled water in a beaker stirring in a water bath at 70 °C. The homogeneous solution was then transferred to a conical flask, which was subsequently ultrasonicated and connected to a vacuum pump for degassing. The stock solutions were kept at 5 °C in a refrigerator. All solutions were prepared with deionized water from a MILLI-Q System (Millpore, MA, USA) and were filtered with 0.20 μm Millipore filters before use.Apparatus
Fused-silica capillaries of 50 μm id and 360 μm od were purchased from Polymicro Technologies (Phoenix, AZ, USA). A CE-L1 (CE Resources, Singapore, Republic of Singapore) or a Spellman CZE 1000R (Plainview, NY, USA) power supply was used to apply high voltage across the capillary. DNA fragments and the EOF marker were detected at 266 nm on a Linear Instrument UVIS 200 (Reno, NV, USA). Electropherograms were recorded and evaluated with a CSW17 software (DataApex, Prague, Czech Republic).Capillary coating and EOF measurement
Capillary coating with linear polyacrylamide was carried out following the method proposed by Hjerten.16 The imidazolium-based IL was covalently bonded onto the capillary surface (Fig. 1) by the following procedures. The fresh capillary was treated for 2 h with 0.5 M sodium hydroxide, 1 h with 1 M hydrochloric acid, 10 min with deionized water, and 10 min with methanol consecutively. It was then heated in a 120 °C oven and flushed gently with pure nitrogen to drive out the residual water and methanol. The Si–O–Si bond was generated on the silica surface by filling the capillary with CPTCS followed by sealing both ends and keeping it at room temperature for 2 h. After that it was rinsed with toluene for 20 min. At room temperature, excess imidazole was dissolved in toluene; the supernatant was filtered and introduced into the capillary by a positive pressure. After it was heated at 90 °C for 6 h, the capillary was rinsed with toluene and dichloromethane successively and subsequently dried with nitrogen under 70 °C for 2 h. It was rinsed with 1-chlorodecane for 10 min then sealed at both ends and heated in oven at 90 °C for 10 h. The pretreated capillary was rinsed successively for 10 min with toluene, 10 min with methanol and 30 min with deionized water before use.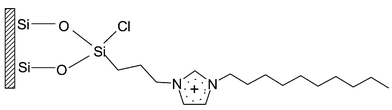 | ||
| Fig. 1 Schematic representation of IL-coated capillary surface. | ||
The EOF of the coated capillary was measured by three consecutive runs. The fast EOF was measured by the migration time of a neutral marker (DMSO solution) from inlet to detector under an electric field. For the low-rate EOF (migration time longer than 30 min), the method described by Williams and Vigh48 was employed.
Results and discussion
EOF of the coated capillary
EOF of the PACC remained cathodic, while EOF of ILCC was reversed due to the bonded imidazolium cations. EOFs in both PACC and ILCC increased cathodically with increasing pH (Table 1) owing to the unshielded surface silanols which would further deprotonate under more alkaline buffer.36| pH | |||||
|---|---|---|---|---|---|
| 4.6 | 6.0 | 7.0 | 7.8 | 9.0 | |
| PACC | 3.4 | 4.4 | 7.1 | 10.4 | 12 |
| ILCC | −6.3 | −3.7 | −3.1 | −1.1 | −0.4 |
Both HEC and PVP added to the buffer will suppress EOF by adsorbing onto the silica surface as well as by increasing the buffer viscosity. However, PVP was found to be more efficient in suppressing EOF. The residual EOFs in the above two capillaries were negligible in the presence of 1% PVP. In the buffer consisting of 1 × TBE and 1% HEC, anodic and cathodic EOFs were observed in ILCC and PACC respectively. PVP is more efficient in suppressing EOF than HEC because it is more hydrophobic.49,50 It was reported that even in a bare silica capillary, the interaction between the hydrophobic PVP and the hydrophobic siloxane on the silica surface favours its adsorption;51 also, as pointed out by Chiari et al.,52 hydrogen bonding between the surface silanol and PVP plays a role in the adsorption and stability of the coating.
Electrophoresis of DNA fragments in free TBE buffer
When electrophoresed in free TBE buffer (without sieving polymer), the components in ΦX174 DNA-Hae III digest mixture comigrated in both ILCC and PACC, and the peak height increased with buffer concentration. All peaks in Fig. 2 are tailing, extenuating with increasing buffer concentration, mainly due to DNA-wall interaction. The electrophoretic behavior of DNA fragments in the two capillaries showed substantial differences. In ILCC, the migration time of the mixture was longer, decreasing with buffer concentration, from 33.3 min in 1 × TBE to 18.3 min in 3 × TBE. While in PACC it was in the range of 8–10 min and slightly increased with buffer concentration. Moreover, the peaks in ILCC were more asymmetric due to the electrostatic interaction between DNA and the positive IL-coating. The above experiments suggest that DNA interacts with both IL- and PA-coated capillaries, while the electrostatic DNA–IL interaction is stronger and is the predominant factor determining the migration time. The cations in TBE buffer compete with the cationic coating for the negatively charged DNA fragments, so the higher the concentration, the weaker the adsorption of DNA onto the IL-coating and therefore the shorter the migration time. The behavior of DNA fragments in the ILCC is similar to that described in capillary electrochromatography (CEC) mode.53 The interaction between DNA and the residual silanol or the coated PA in the PACC may be attributed to hydrogen bonding or hydrophobic adsorption, which is weaker compared to the electrostatic attraction. Although high-concentration buffer helps to elute the fragments from the surface (which can be confirmed from the increasing peak heights), the high ionic strength will also lead to the reduced double layer thickness and increased buffer viscosity and thereby low migration velocities of the analytes. The migration of DNA fragments in PACC is more CZE-characterized.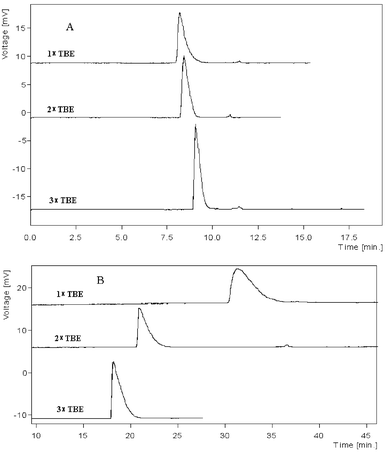 | ||
| Fig. 2 Electropherograms of DNA in ILCC and PACC A: PACC; B:ILCC. Running buffer: TBE with concentrations stated in the figure. Length of ILCC and PACC: 50/38.5 cm (total/effect length). Concentration of the DNA: ΦX174 DNA-Hae III digest diluted to 40.4 μg ml−1. Sample injection: 3 kV × 3 s. Applied voltage: −15 kV. UV detection was set at 266 nm. | ||
Electrophoresis of DNA fragments in sieving matrix
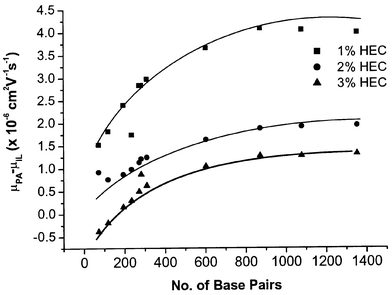 | ||
| Fig. 3 Dependence of DNA–IL interaction on number of base pairs. Buffer: 2 × TBE plus desired concentration of HEC as stated in the figure. Other conditions as in Fig. 2. | ||
Fig. 3 also illustrates that the mobility difference decreases with increasing concentration of sieving matrix, suggesting that the capillary wall was further covered by the increasing amount of HEC added into the buffer thus the interaction between DNA fragments and IL-cations became weaker. Moreover, the collision frequency between DNA and the wall would decrease due to the high concentration sieving polymer.54
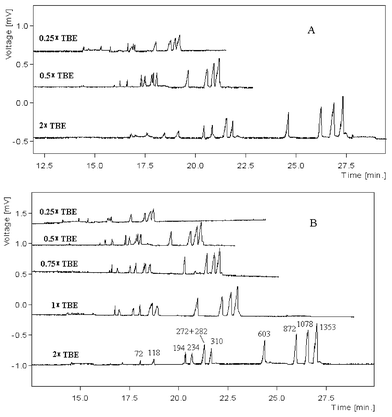
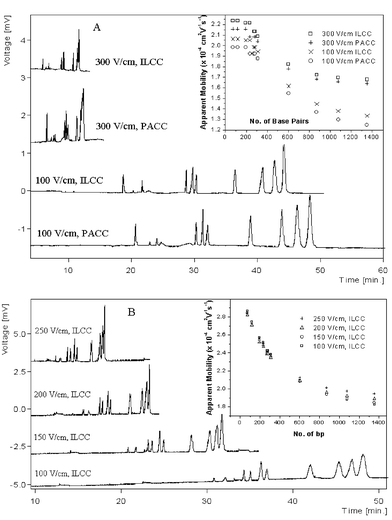 | ||
| Fig. 5 Influence of electric field strength. Buffer: A, 2 × TBE + 2% HEC; B, 2 × TBE + 2% PVP. The experiments were conducted in ILCC. | ||
Stability of the IL-coating
The stability of the IL- and PA-coating was investigated in a 4 day period with two runs a day. 2% HEC was chosen as sieving matrix because it could separate the DNA fragments (although partially, but enough for performance assessment) and did not completely cover the coating surface. Every day in the morning, the ΦX174 standard was electrophoresed in the fresh buffer; eight hours later, another run was carried out. The buffer was left in the capillary over night. The migration times of the fragments showed detectable trends in the two capillaries, which suggests the gradual hydrolysis of the Si–O–Si bond. But the change was very slow and the maximum relative errors in migration time were 4.78% for ILCC and 3.71% for PACC (Table 2): however plate number and resolution between peaks did not decrease significantly, suggesting that the deterioration of the capillary coating was not so serious. Considering the capillary effective length, the separation efficiencies were all more than 105 plates m−1. The ILCC can be used with the separation buffer for at least 96 h without substantial deterioration in performance.| Migration time/min | Plate number (×104) | Resolution | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ILCC | PACC | ILCC | PACC | ILCC | PACC | |||||||
| No. of bp | 1st | 96th | 1st | 96th | 1st | 96th | 1st | 96th | 1st | 96th | 1st | 96th |
| a in ILCC, the peaks of 72 bp and 118 bp merged | ||||||||||||
| 72 | 22.89 | 23.69 | 11.4 | 19.7 | ||||||||
| 118 | 21.77a | 22.81 | 23.96 | 24.71 | 33.0 | 38.8 | 12.4 | 11.4 | 3.9 | 3.8 | ||
| 194 + 234 | 28.85 | 29.52 | 30.22 | 31.07 | 22.0 | 20.5 | 14.5 | 11.6 | 46.3 | 47.9 | 21.3 | 22.5 |
| 271 + 281 | 29.81 | 30.50 | 31.23 | 31.91 | 8.6 | 7.9 | 9.4 | 10.2 | 3.1 | 2.7 | 2.8 | 2.8 |
| 310 | 30.46 | 31.21 | 31.99 | 32.71 | 13.8 | 10.7 | 9.9 | 13.2 | 1.8 | 2.1 | 1.9 | 2.0 |
| 603 | 36.65 | 37.71 | 38.84 | 39.80 | 6.7 | 7.0 | 7.0 | 6.9 | 13.8 | 13.6 | 13.7 | 13.4 |
| 872 | 40.93 | 42.30 | 43.77 | 44.94 | 4.3 | 4.5 | 5.0 | 5.9 | 6.3 | 7.0 | 7.2 | 7.4 |
| 1078 | 42.93 | 44.51 | 46.21 | 47.52 | 4.6 | 4.1 | 4.6 | 6.1 | 2.5 | 2.5 | 3.0 | 3.1 |
| 1353 | 44.37 | 46.20 | 48.19 | 49.98 | 6.8 | 7.3 | 4.8 | 4.2 | 1.9 | 1.8 | 2.3 | 2.5 |
ILCC showed strong interactions with DNA fragments in buffers of low ionic strengths because of their opposite charges. The DNA–IL interaction is determined by the electrostatic attraction that increases with fragment charge density; the DNA fragment with higher charge density was more retarded. With the introduction of a sieving polymer, DNA fragments can also be separated in ILCC with similar patterns to that of PACC. With the anodic EOF, the ILCC can offer shorter analysis time than PACC does in the presence of the often-used weak self-coating polymer. This is potentially a significant advantage of ILCC in separating large-size DNA fragments. Furthermore, ILCC may be useful in the separation of other anionic species under the IE-OTCEC mode. The ILCC exhibits comparable long-term stability as the PACC in DNA separation.
Acknowledgements
The authors thank the National University of Singapore for financial support for the research presented in the paper.References
- V. Dolnik, J. Biochem. Biophys. Methods, 1999, 41, 103 CrossRef CAS.
- S. M. Clark and R. A. Mathies, Anal. Chem., 1997, 69, 1355 CrossRef CAS.
- Y. Fang, J. Z. Zhang, J. Y. Hou, H. Lu and N. J. Dovichi, Electrophoresis, 1996, 17, 1436 CAS.
- D. N. Heiger, A. S. Cohen and B. L. Karger, J. Chromatogr., 1990, 516, 33 CrossRef CAS.
- M. Chiari, M. Cretich and J. Horvath, Electrophoresis, 2000, 21, 1521 CrossRef CAS.
- N. C. Stellwagen, C. Gelfi and P. G. Righetti, Biopolymers, 1997, 42, 687 CrossRef CAS.
- M. Chiari, S. Riva, A. Gelain, A. Vitale and E. Turati, J. Chromatogr., A, 1997, 781, 347 CrossRef CAS.
- A. E. Barron, W. M. Sunada and H. W. Blanch, Electrophoresis, 1996, 17, 744 CAS.
- D. T. Mao, J. D. Levin, L. Y. Yu and R. M. A. Lautamo, J. Chromatogr., B, 1998, 714, 21 CrossRef CAS.
- F. T. Han, B. H. Huynh, Y. F. Ma and B. C. Lin, Anal. Chem., 1999, 71, 2385 CrossRef CAS.
- R. S. Madabhushi, M. Vainer, V. Dolnik, S. Enad, D. L. Barker, D. W. Harris and E. S. Mansfield, Electrophoresis, 1997, 18, 104 CAS.
- C. H. Wu, T. B. Liu and B. Chu, Electrophoresis, 1998, 19, 231 CAS.
- H. S. Chen and H. T. Chang, Anal. Chem., 1999, 71, 2033 CrossRef CAS.
- H. M. Pang, V. Pavski and E. S. Yeung, J. Biochem. Biophys. Methods, 1999, 41, 121 CrossRef CAS.
- J. Horvath and V. Dolnik, Electrophoresis, 2001, 22, 644 CrossRef CAS.
- S. Hjerten, J. Chromatogr., 1985, 347, 191 CrossRef.
- H. J. Tian, L. C. Brody, D. Mao and J. P. Landers, Anal. Chem., 2000, 72, 5483 CrossRef CAS.
- K. Srinivasan, C. Pohl and N. Avdalovic, Anal. Chem., 1997, 69, 2798 CrossRef CAS.
- M. Nakatani, A. Shibukawa and T. Nakagawa, Electrophoresis, 1995, 16, 1451 CAS.
- N. J. Munro, A. F. R. Huhmer and J. P. Landers, Anal. Chem., 2001, 73, 1784 CrossRef CAS.
- C. Gelfi, M. Curcio, P. G. Righetti, R. Sebastiano, A. Citterio, H. Ahmadzadeh and N. J. Dovichi, Electrophoresis, 1998, 19, 1677 CAS.
- M. X. Huang, J. Plocek and M. V. Novotny, Electrophoresis, 1995, 16, 396 CAS.
- M. Chiari, M. Nesi, J. E. Sandoval and J. J. Pesek, J. Chromatogr., 1995, 717, 1 CrossRef CAS.
- J. T. Smith and Z. El Rassi, Electrophoresis, 1993, 14, 396 CAS.
- D. Schmalzing, C. A. Piggee, F. Foret, E. Carrilho and B. L. Karger, J. Chromatogr., A, 1993, 652, 149 CrossRef CAS.
- Z. X. Zhao, A. Malik and A. L. Lee, Anal. Chem., 1993, 65, 2747 CrossRef CAS.
- S. Hjerten and K. Kubo, Electrophoresis, 1993, 14, 390 CAS.
- A. Cifuentes, M. Defrutos, J. M. Santos and J. C. Diezmasa, J. Chromatogr., 1993, 655, 63 CrossRef CAS.
- M. A. Strege and A. L. Lagu, J. Chromatogr., 1993, 630, 337 CrossRef.
- J. K. Towns, J. M. Bao and F. E. Regnier, J. Chromatogr., 1992, 599, 227 CrossRef CAS.
- W. Nashabeh and Z. El Rassi, J. Chromatogr., 1991, 559, 367 CrossRef CAS.
- K. A. Cobb, V. Dolnik and M. Novotny, Anal. Chem., 1990, 62, 2478 CrossRef CAS.
- H. J. Jung and Y. C. Bae, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1405 Search PubMed.
- M. F. Huang, C. E. Hsu, W. L. Tseng, Y. C. Lin and H. T. Chang, Electrophoresis, 2001, 22, 2281 CrossRef CAS.
- H. S. Chen and H. T. Chang, J. Chromatogr., A, 1999, 853, 337 CrossRef CAS.
- C. Y. Liu, Y. W. Ho and Y. F. Pai, J. Chromatogr., A, 2000, 897, 383 CrossRef CAS.
- J. S. Fritz, M. C. Breadmore, E. F. Hilder and P. R. Haddad, J. Chromatogr., A, 2002, 942, 11 CrossRef CAS.
- M. A. Rehder and L. B. McGrown, Electrophoresis, 2001, 22, 3759 CrossRef CAS.
- R. Xiang and C. Horváth, Anal. Chem., 2002, 74, 762 CrossRef CAS.
- C. J. van Oss, Colloids Surf., A, 1993, 78, 1 CrossRef CAS.
- K. M. Dieter, C. J. Dymek, N. E. Heimer, J. W. Rovang and J. S. Wilkes, J. Am. Chem. Soc., 1988, 110, 2722 CrossRef CAS.
- S. Dai, Y. H. Ju and C. E. Barnes, J. Chem. Soc., Dalton Trans., 1999, 1201 RSC.
- D. W. Armstrong, L. F. He and Y. S. Liu, Anal. Chem., 1999, 71, 3873 CrossRef CAS.
- S. Chun, S. V. Dzyuba and R. A. Bartsch, Anal. Chem., 2001, 73, 3737 CrossRef CAS.
- M. Vaher, M. Koel and M. Kaljurand, Chromatographia, 2001, 53, S302 Search PubMed.
- E. G. Yanes, S. R Gratz, M. J. Baldwin, S. E. Robison and A. M. Stalcup, Anal. Chem., 2001, 73, 3838 CrossRef CAS.
- M. H. Valkenberg, C. Decastro and W. F. Holderich, Top Catal., 2001, 14, 139 Search PubMed.
- B. A. Williams and G. Vigh, Anal. Chem., 1996, 68, 1174 CrossRef CAS.
- M. N. Albarghouthi, B. A. Buchholz, P. J. Huiberts, T. M. Stein and A. E. Barron, Electrophoresis, 2002, 23, 1429 Search PubMed.
- R. S. Madabhushi, Electrophoresis, 1998, 19, 224 CAS.
- T. Tanahashi, M. Kawaguchi, T. Honda and A. Takahashi, Macromolecules, 1994, 27, 606 CrossRef CAS.
- M. Chiari, M. Cretich, F. Damin, L. Ceriotti and R. Consonni, Electrophoresis, 2000, 21, 909 CrossRef CAS.
- M. C. Breadmore, E. F. Hilder, M. Macka, N. Avdalovic and P. R. Haddad, Electrophoresis, 2001, 22, 503 CrossRef CAS.
- Y. Jin, B. C. Lin and Y. S. Fung, Electrophoresis, 2001, 22, 2150 CrossRef CAS.
- N. C. Stellwagen, A. Bossi, C. Gelfi and P. G. Righetti, Anal. Biochem., 2000, 287, 167 CrossRef CAS.
- C. Heller, Electrophoresis, 1998, 19, 3114 CAS.
| This journal is © The Royal Society of Chemistry 2003 |