Analysis of hydroponic fertilizer matrixes for perchlorate: comparison of analytical techniques†
Timothy W.
Collette
*a,
Ted L.
Williams
a,
Edward T.
Urbansky
*b,
Matthew L.
Magnuson
b,
Gretchen N.
Hebert
c and
Steven H.
Strauss
*c
aUnited States Environmental Protection Agency, Office of Research and Development, National Exposure Research Laboratory, 960 College Station Road, Athens, GA 30605, USA. E-mail: collette.tim@epa.gov
bUnited States Environmental Protection Agency, Office of Research and Development, National Risk Management Research Laboratory, 26 West Martin Luther King Drive, Cincinnati, OH 45268, USA. E-mail: urbansky.edward@epa.gov
cDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523, USA. E-mail: strauss@lamar.colostate.edu
First published on 13th December 2002
Abstract
Seven retail hydroponic nitrate fertilizer products, two liquid and five solid, were comparatively analyzed for the perchlorate anion (ClO4−) by ion chromatography (IC) with suppressed conductivity detection, complexation electrospray ionization mass spectrometry (cESI-MS), normal Raman spectroscopy, and infrared spectroscopy using an attenuated total reflectance crystal (ATR-FTIR) coated with a thin film of an organometallic ion-exchange compound. Three of the five solid products were found by all techniques to contain perchlorate at the level of approximately 100–350 mg kg−1. The remaining products did not contain perchlorate above the detection level of any of the techniques. Comparative analysis using several analytical techniques that depend on different properties of perchlorate allow for a high degree of certainty in both the qualitative and quantitative determinations. This proved particularly useful for these samples, due to the complexity of the matrix. Analyses of this type, including multiple spectroscopic confirmations, may also be useful for other complicated matrixes (e.g., biological samples) or in forensic/regulatory frameworks where data are likely to be challenged. While the source of perchlorate in these hydroponic products is not known, the perchlorate-to-nitrate concentration ratio (w/w) in the aqueous extracts is generally consistent with the historical weight percent of water soluble components in caliche, a nitrate-bearing ore found predominantly in Chile. This ore, which is the only well-established natural source of perchlorate, is mined and used, albeit minimally, as a nitrogen source in some fertilizer products.
Introduction
Perchlorate (ClO4−) has been identified, beginning in the late 1990’s, as a contaminant in some natural waterways that are used extensively for recreation, drinking, and crop irrigation. Concern over perchlorate arises from its ability to interfere with the function of the thyroid,1 which can affect metabolism and development. Based on the US Environmental Protection Agency’s (EPA) latest risk assessment draft,2 a concentration near 1 ppb is presumed to be safe for drinking water.3 The EPA added perchlorate to the Contaminant Candidate List for drinking water in 1998,4 and to the Unregulated Contaminant Monitoring Regulation list in 1999.5 This has led to the development and application of a variety of techniques and methods for determining perchlorate in environmental samples, especially finished drinking water and the ground and surface waters that are used as drinking water sources. These include ion chromatography (IC),6–8 complexation electrospray ionization mass spectrometry (cESI-MS),9–12 and high field asymmetric waveform mass spectrometry (FAIMS).13,14 With suitable sample clean-up, some of these techniques have also been applied to the analysis of aqueous extracts of plants that could absorb and accumulate perchlorate, e.g., salt cedar,15 fruits and vegetables,16 and tobacco.17Perchlorate contamination, which occurs mostly in the Western USA, can be attributed primarily to the defense and aerospace industry or to military operations,11,18 where salts of perchlorate are used as oxidants in solid fuels for rockets and munitions. However, to properly assess the significance of perchlorate contamination, and to minimize its future impact, inquiries should be made into the possibility of additional sources of loading to the environment and to the food supply.
The only consistent and well-established natural occurrence of perchlorate is in nitrate-bearing ores, called caliche, that are located predominantly in Chile.19–22 These ores are mined, and NaNO3 (Chile saltpeter) is refined and sold as a finished fertilizer product (N–P–K grade 16–0–0, i.e., 16% N, 0% P2O5, 0% K2O by mass). Also, Chile saltpeter may be used infrequently in the manufacture of other fertilizer products, including some soluble plant foods that are sold to consumers at nurseries, home improvement centers, and other retail stores. Furthermore, KNO3 made from Chile saltpeter by cation exchange is sold as a finished fertilizer product (N–P–K grade 14–0–45) and may similarly be incorporated into some soluble plant foods.
It is well-known that salts of perchlorate are carried-over with NaNO3 in the process of refining Chile saltpeter. Therefore, products derived from Chilean caliche contain perchlorate as a minor component. For example, the finished 16-0-0 fertilizer product acquired in the last few years has been found to consistently contain perchlorate just below the level of 2000 mg kg−1.17,20,23 The only current vendor for this material has recently modified the caliche refinement process in order to reduce the level of perchlorate to less than 100 mg kg−1.24
Fertilizer products derived from Chilean ores were widely used many decades ago. However, their usage has diminished to less than 0.2% of current USA fertilizer consumption,25 due primarily to the low cost of synthetically produced nitrogen sources. Thus, it was surprising when reports of perchlorate occurrence in certain lawn-and-garden fertilizer products with no known link to Chile saltpeter, which were purchased from November 1998 to January 1999, began to appear (see Williams et al.23 and references therein). While the initial work26 on this topic relied mainly on IC for many of the materials tested, both it and later confirmatory works23,27 included analysis of some samples by capillary electrophoresis, Raman spectroscopy, and NMR spectroscopy. Applying methods to confirm IC results with complicated matrixes, such as fertilizers, is important to prevent false positives (there are many unknown ions that might co-elute) as well as false negatives (there are many factors that raise detection limits). The utilities of Raman spectroscopy23 and cESI-MS28 in this regard have been demonstrated recently. In addition to describing confirmatory analytical techniques, these two recent studies23,28 report on the absence or presence of perchlorate in approximately 70 fertilizer materials with no known link to Chile saltpeter that were acquired after May 1999. From among these approximately 70 materials, which span a wide range of type, perchlorate was detected in only two; both of these were fertilizer products formulated specifically for use in hydroponics.23 (These two hydroponic fertilizers are a subset of the seven products analyzed in this work.)
EPA recently released a report 25 on the most comprehensive survey to date of the presence or absence of perchlorate in fertilizers and related materials. Samples were acquired in May 2000 and included products from all major national suppliers of large-scale agriculturally relevant materials. The survey also included some finished lawn-and-garden fertilizers, but did not include fertilizers targeted specifically for hydroponic use. Perchlorate was not found in any of the approximately 50 tested materials except for the few known to originate from Chile saltpeter.
It is important to note that all surveys of fertilizers for perchlorate represent only a temporal snapshot defined by the time at which samples were acquired. Nonetheless, there is clear consensus among researchers that currently used fertilizers are negligible contributors to perchlorate in the environment, based on the weight of evidence to date23,25,28,29 and on consideration of the low usage of products derived from Chilean caliche. The source of perchlorate in some lawn-and-garden fertilizers acquired from November 1998 to January 1999,23 and in the hydroponic fertilizer products described in this report (which were acquired during the Spring of 2000), remains unknown.
Apart from consideration of the magnitude of environmental loading, the finding of perchlorate in hydroponic fertilizers raises some particular concerns. To understand these concerns, note that hydroponics is defined as the production of crops without soil. This includes both water-based and media-based (e.g., Rockwool) systems. Further note that hydroponic crop production is usually conducted in a greenhouse environment that supplies all water and nutrients, which are often re-circulated.30
If plants grown conventionally in soil are treated with perchlorate-containing fertilizers, the amount of perchlorate available to the plant will likely be attenuated over time via several mechanisms, including runoff and dilution due to rainfall, adsorption to soil, and biodegradation by soil microbes. On the other hand, hydroponic cultures seem more likely to experience accumulation of perchlorate within the media while maintaining bioavailability to growing plants since these natural attenuation mechanisms are absent. Furthermore, all perchlorate-reducing microbes identified to date favor nitrate over perchlorate as a terminal oxidant (electron acceptor) for metabolism.31–35 In contrast to fertilizers for soil-based farming, nitrate is the primary source of nitrogen in hydroponic fertilizers, largely due to the high water solubility of its salts. Consequently, it is unlikely that significant consumption of perchlorate by water-borne microbes will be observed in hydroponic cultures where there normally would be excess nitrate ion present as a nutrient. This assertion is supported by recent investigations on the removal of perchlorate from water by willow trees grown in sand and hydroponic bioreactors,36 using nutrient solutions containing nitrate at levels consistent with those used in hydroponic crop production.37
However, it should not be inferred that mere exposure of a plant to perchlorate in an aqueous hydroponic medium (via root or foliage) means that perchlorate absorption or accumulation definitely occurs. For example, absorption can be affected by a variety of factors, including the presence and concentrations of other ions in the medium, the types of transport proteins involved in the process, and species-, variety-, or cultivar-specific properties of the plant. None of these variables have yet been studied sufficiently in terms of their effects on perchlorate uptake. In addition, many plants can accumulate xylem-transportable ions in their stems or foliage, but not in their fruits, seeds, or nuts. Indeed, transformation of perchlorate (i.e., reduction) has been shown to occur within the tissue of some plants following absorption.38 Consequently, the focus of agronomic and botanical studies would have to be on uptake in the edible portions of food plants before implications for human or animal health can be inferred.
Although hydroponically grown crops constitute a small percentage of the total global market, the worldwide commercial hydroponics industry continues to grow steadily—about four-fold in known area harvested from 1990 to 2001.30 Many developed western countries have some commercial hydroponic operations, and the industry is quite large and well-established in, for example, The Netherlands, United Kingdom, Spain, Canada, and Japan.30,37,39,40 Indeed, a significant percentage of the global supply of a few fresh-market commercial fruits and vegetables (e.g., tomatoes, cucumbers, capsicums, and lettuce) are now grown hydroponically. This increase in commercial hydroponic farming is driven by practical advantages that are perceived both by some producers (e.g., higher and more consistent yield) and by some consumers (e.g., better taste and appearance). Also, hydroponics is viewed by some as environmentally friendly since water is often recycled and pesticide use is often limited. Furthermore, hydroponic farming is attractive to countries where arable land and water are scarce (e.g., Israel).41 Apart from large-scale commercial businesses, hydroponics is also increasingly attractive for other horticultural activities such as hobby gardening and urban agriculture. The increase in hydroponic activity in all sectors is expected to continue into the foreseeable future.30
Therefore, in response to this situation, we have analyzed a small set of widely available hydroponic fertilizers, and herein report our results. Our goal was not to present an exhaustive survey of all such products or to comment on general occurrence. Instead, we sought to illuminate this potential exposure route, and to illustrate, for the first time, the benefits of jointly employing certain spectroscopic techniques that depend on different properties of perchlorate in order to confirm chromatographic identifications in complex matrixes.
Experimental
Sample procurement and preparation
Hydroponic fertilizers were purchased at a retail store in Athens, GA, during March and May of 2000 (see Table 1). To obtain a reasonably representative sample, the complete contents of a solid fertilizer (samples #1–5 of Table 1) container, ranging in weight from about 225–675 g, was riffled into two halves with one of these halves being riffled again; riffling of a subsequent half was repeated until less than 75 g of material was obtained in a half. The final riffled material was then placed in a large bottle where it was further mixed via rolling and shaking of the bottle. Next, the material was divided into three parts. One part was retained in Athens, GA, for analysis by Raman spectroscopy, one part sent to Cincinnati, OH, for analysis by IC and cESI-MS, and one part sent to Fort Collins, CO, for analysis by ATR-FTIR spectroscopy. Extracts of these solid fertilizers were then prepared under somewhat different conditions at each laboratory as described below, reflecting different instrument requirements. In particular, the solid-to-water ratio for the extracts varied considerably depending on the demands of the various techniques. The liquid fertilizer (samples #6 and #7 of Table 1) containers were shaken for at least 2 min and the contents were likewise divided among the three laboratories for analysis. All samples were stored in sealed amber glass bottles for overnight shipping from Athens, GA, to the other laboratories.| ClO4− concentration/mg kg−1c | ||||||
|---|---|---|---|---|---|---|
| Sample #a | N–P–K grade | Purchase dateb | IC | cESI-MS | ATR-FTIR | Raman |
| a Samples 1–5 were solid fertilizers. Samples 6 and 7 were liquid fertilizers. b All fertilizers were purchased at a retail store in Athens, GA, USA. c Perchlorate concentration in product as purchased. Value is mean ± sample standard deviation. ND denotes that perchlorate was not detected. As discussed in the text, detection levels differ for each technique and for each matrix, but ND for all techniques can be interpreted as perchlorate <30 mg kg−1. Note: These results do not necessarily represent a continuing level of perchlorate in these or other fertilizer products. (See text for elaboration.) | ||||||
| 1 | 20–6–16 | 3–2–00 | 202 ± 2 | 230 ± 29 | 120 ± 40 | 92 ± 5 |
| 2 | 5–18–22 | 3–2–00 | 319 ± 16 | 483 ± 16 | 340 ± 30 | 352 ± 87 |
| 3 | 15.5–0–0 | 3–2–00 | ND | ND | ND | ND |
| 4 | 9–5–18 | 5–22–00 | 285 ± 9 | 323 ± 15 | 300 ± 100 | 268 ± 12 |
| 5 | 15–0–0 | 5–22–00 | ND | ND | ND | ND |
| 6 | 7–0–5 | 5–22–00 | ND | ND | ND | ND |
| 7 | 5–2–6 | 5–22–00 | ND | ND | ND | ND |
Note that the concentrations of perchlorate and nitrate reported in Tables 1 and 2, respectively, assume complete extraction of water-soluble analytes from the solid matrixes in all cases. This has not been independently established. Indeed, 100% extraction may not be achieved for all solid fertilizer matrixes, particularly when high solid-to-water ratios are used and some undissolved solids remain, as is the case with some extracts for Raman determination of perchlorate. However, reasonable quantitative agreement among all techniques (see Tables 1 and 2) is consistent with near-complete extraction. Nonetheless, concentrations reported here for solid products should be interpreted as approximate since matrix-specific extraction efficiencies are not accurately known.
| Samplea | % Nitrate nitro gen as listedb | % Nitrate nitro gen Ramanc | % Nitrate nitro gen ATR-FTIRc | ClO4−/NO3− w/w ratio Ramand | ClO4−/NO3− w/w ratio ATR-FTIRd |
|---|---|---|---|---|---|
| a #1–#7 refer to samples listed in Table 1. Bulldog Soda is sodium nitrate derived solely from mined Chilean caliche. The Raman and ATR-FTIR measured values for perchlorate in Bulldog Soda are 1759 ± 111 and 2200 ± 300 mg kg−1, respectively. b The weight % nitrate nitrogen in the fertilizer as reported by the manufacturer on the product package. c The weight % nitrate nitrogen in the fertilizer as measured by Raman spectroscopy or by ATR-FTIR spectroscopy. d The perchlorate-to-nitrate concentration ratio (w/w), as determined by Raman spectroscopy or by ATR-FTIR spectroscopy. If this ratio is in or near the range of 3.7 × 10−4 to 9.0 × 10−2, the level of perchlorate relative to nitrate is consistent with products derived from mined Chile saltpeter at the time these products were purchased. See text for details. N/A denotes not applicable since perchlorate was not detected. | |||||
| Bulldog Soda | 16.0 | 16.4 ± 0.3 | 18.1 ± 0.9 | (2.4 ± 0.2) × 10−3 | (2.8 ± 0.4) × 10−3 |
| #1 | 12.5 | 12.6 ± 0.1 | 14.5 ± 0.7 | (1.7 ± 0.1) × 10−4 | (1.9 ± 0.5) × 10−4 |
| #2 | 5.0 | 3.3 ± 0.1 | 4.1 ± 0.2 | (2.4 ± 0.6) × 10−3 | (1.9 ± 0.2) × 10−3 |
| #3 | 15.5 | 14.1 ± 0.1 | 15.6 ± 0.7 | N/A | N/A |
| #4 | 8.9 | 7.5 ± 0.1 | 8.1 ± 0.5 | (8.1 ± 0.5) × 10−4 | (8.0 ± 3.0) × 10−4 |
| #5 | 15.0 | 14.5 ± 0.1 | 16.7 ± 0.9 | N/A | N/A |
| #6 | 7.0 | 6.2 ± 0.1 | 7.2 ± 0.5 | N/A | N/A |
| #7 | 4.55 | 3.7 ± 0.1 | 4.3 ± 0.2 | N/A | N/A |
Ion chromatography (IC)
A solution of each solid fertilizer product was prepared by dissolving the solid into deionized (DI) water at a ratio of 1.00 ± 0.05 g dL−1 (∼1% w/w). The aqueous solutions were injected (100 μL loop) into a Dionex (Sunnyvale, CA) DX 300 ion chromatograph equipped with IonPac AG16 (guard) and AS16 (analytical separation) columns. The eluent was 0.100 M NaOH(aq) solution at a flow rate of 1.0 mL min−1. Detection was by suppressed conductometry with a CDM2 conductivity detector; the current in the ASRS Ultra suppressor was set to 300 mA. The instrument was calibrated from 0.050 to 100 μg mL−1 perchlorate using multiple NaClO4(aq) standards prepared from a stock solution of the reagent grade salt (GFS Chemical, Columbus, OH) using water deionized in-house by reverse osmosis. Water used for the eluent and the suppressor, which was operated in the external water mode, was obtained from a Waters (Milford, MA) Milli-Q polishing unit. Sodium hydroxide (50% w/w) used to make the eluent was obtained from Fisher Scientific (Pittsburgh, PA).Under these conditions, the concentration of the bulk fertilizer constituent anions (i.e., nitrate, sulfate, phosphate, and chloride) exceeded the perchlorate concentration by at least several hundredfold. Although these other anions are essentially not retained by the AS16 column, their combined concentration is so great that they produce a large, tailing peak. The peak of perchlorate, which has a retention time of ∼8.4 min, sits on this tail, as shown in Fig. 1a. Fortifications of 2 μg mL−1 were used to verify the identity of the perchlorate peak as illustrated in Fig. 1b; this concentration was chosen so as to roughly double the peak area (i.e., about twice the analyte concentration found in unspiked samples).
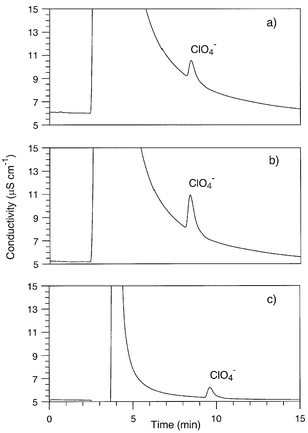 | ||
| Fig. 1 (a) Ion chromatogram of a 1.0 g dL−1 solution of fertilizer sample #1 listed in Table 1. Conditions: 0.10 M NaOH(aq) as eluent; flow rate: 1.0 mL min−1; AG16/AS16 columns; 100 μL loop; suppressed conductivity detection. (b) Same after fortification with 2.0 μg mL−1 perchlorate. (c) Ion chromatogram of the 8.0–10.0 min eluate fraction of the sample whose chromatogram is (a). The fraction was reinjected onto the IC using a 1.0 mL sample loop; otherwise, conditions are the same. | ||
Complexation electrospray ionization mass spectrometry (cESI-MS) combined with IC eluate fraction collection
Perchlorate-containing fractions of the IC eluate were collected and analyzed by cESI-MS. (The very complex matrix of these samples interferes with effective cESI-MS analysis when the technique of standard addition9,10,28 is used without prior anion isolation.) In this fashion, each injected aliquot is analyzed both by conductivity and, subsequently, by mass spectrometry. An increase in selectivity is afforded with the combined use of retention time (from IC) and mass-to-charge ratio (from cESI-MS). Unfortunately, analyte concentration is reduced because of dilution during fraction collection. However, the dilution factor was not prohibitive due to the high sensitivity of the cESI-MS technique.IC fractions eluting between 8.0 and 10.0 min (2.0 mL total per fraction) were collected for multiple injections and subsequently combined using a Gilson (Middleton, WI) model FC 203B fraction collector. (The perchlorate peak elutes over the course of ∼1.5 min with 0.10 M NaOH eluent.) With more pristine samples, the analyte-free post-suppressor eluate from the IC can be collected and used successfully as a blank for cESI-MS analyses of IC fractions. However, for most fertilizer solutions the post-suppressor eluate does not constitute a useful blank with these IC fractions because the perchlorate peak elutes on the large tail of the bulk nutrient anions peak. These anions reduce the efficiency of the electrospray process, adversely affecting the background mass spectrometer signal. Therefore, to further reduce the level of these anions, the combined IC eluate fractions were reinjected on the IC using a 1.00 mL loop, and multiple fractions were collected/combined a second time.
The large injection volume used in the second set of collections resulted in some broadening of the perchlorate peak and also increased its retention time to ∼9.6 min, as verified by injecting a standard solution. Accordingly, the eluate was taken during the period 9.0–11.0 min. As illustrated in Fig. 1c, the baseline conductivity (L) of the chromatogram for the second set of collections (L = 5.3 μS cm−1) was significantly less than that for the first set (L > 8.0 μS cm−1, see Fig. 1a). For these fertilizer samples, the combined fractions gathered from the second set of collections proved adequate, and were subsequently used in the cESI-MS analyses described below.
To prepare the IC eluate for cESI-MS analysis, a 1.0 mL aliquot of the collected eluate was transferred to a 4.0 mL glass vial using a pipettor. The following were then added to the eluate: 30 μL 0.20 M C10H21NMe3Br(aq) (Fluka, Buchs, Switzerland) and 500 μL CH2Cl2 (Fisher Optima). The vial was capped with a PTFE-lined septum and shaken for 90 s. After phase separation (2–5 min), the dichloromethane layer was drawn off with a gas-tight syringe; a volume of ∼350 μL was recovered. This dichloromethane extract was injected into the electrospray interface with a Rheodyne (Rohnert Park, CA) model 7725 injector with a 200 μL loop. The carrier liquid, Optima grade methanol, was pumped at 0.3 mL min−1 by a Waters 600 pump (Milford, MA). The mass spectrometer was a Finnigan MAT TSQ-700 (San Jose, CA) equipped with a standard Finnigan electrospray interface. Mass spectra were acquired in the negative ion mode by scanning Q3 over appropriate mass ranges with a scan time of 0.5 s. The ESI spray voltage was 4.0 kV, the interface capillary temperature was 200 °C, and the sheath gas pressure was 70 psi (480 kPa).
Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy
Dichloromethane (Fisher, Fairlawn, NJ) and lithium perchlorate (LiClO4, GFS Chemical, Columbus, OH) were obtained and used without further purification. The polyalkylated ferrocenium salt 1,1′,3,3′-tetrakis(2-methyl-2-nonyl)ferrocenium nitrate (DEC+NO3−) was synthesized according to a literature method.42,43 This compound is one of a number of selective, ferrocene-based, water-insoluble, organometallic ion-exchange compounds.42–46 All aqueous stock solutions were prepared in Class A volumetric glassware using distilled DI water (Barnstead NANOpure, Dubuque, IA) that had an initial resistivity of 18 MΩ cm. All experiments were performed at 24 ± 1 °C. The seven fertilizer samples were dissolved in water to make 10 g L−1 stock solutions of all samples except for #4 for which a 3 g L−1 stock solution was made.The spectrometer used in all experiments was an ASI (Applied Systems Inc, Millersville, MD) ReactIR™-1000 equipped with an ASI SiComp® ATR-FTIR immersion probe. The spectrometer was equipped with a liquid-nitrogen-cooled MCT detector. The spectral window was 4000–650 cm−1 with a nominal spectral resolution of 8 cm−1. Happ-Ganzel apodization was used with no post-run spectral smoothing. Data collection and manipulation was carried out using ASI ReactIR™ software (version 2.1). The probe consisted of a 30-bounce silicon ATR crystal mated to a ZnSe optical focusing element and housed in a 1.5 in. long × 1 in. diameter cylindrical stainless-steel conduit. The exposed surface of the ATR crystal was a circular area 1 cm in diameter. For some experiments, the immersion probe was inverted so that a small amount of aqueous solution could wet the entire surface of the crystal. In other experiments, the probe was immersed in a beaker containing 100 mL of aqueous sample.
Initial characterization of the fertilizer samples was accomplished by placing a sufficiently large aliquot of the stock solution on top of the inverted SiComp® probe so that the ATR crystal was completely covered by the sample (this was generally 1 mL) and collecting a spectrum (64 co-added scans; 35 s total collection time) that was ratioed to DI water. The absorbance (A) of the ν3 peak of nitrate at 1347 cm−1, which is due to the asymmetric NO3 stretching vibration,47 was used in these experiments to determine the concentration of nitrate in each 10 or 3 g L−1 fertilizer stock solution. This was done by using a calibration curve of A(1347 cm−1) vs. nitrate concentration, which was found to be linear over the concentration range 0.62 g L−1 to 25 g L−1 (a similar linear calibration curve has been reported in the literature).48 The ν3 peak of perchlorate in water at 1104 cm−1, which is due to the asymmetric ClO4 stretching vibration,47 was not observed in any of the stock solutions, indicating that the concentration of perchlorate in the samples was less than 230 ppm (see below).
The LOD of perchlorate in DI water using the SiComp® probe was found to be 230 ppm, which is more than four orders of magnitude higher than early estimates of a safe level for drinking water (4–18 ppb).18 In this case, the LOD is defined as the concentration of perchlorate that results in an absorbance at 1104 cm−1 after 15 min (3750 co-added scans) that is three times the signal to noise ratio (S/N) at ∼1100 cm−1. To lower the perchlorate LOD of the ATR-FTIR spectrometer, the silicon ATR crystal was coated with a thin film of DEC+NO3−. Some aspects of this procedure have been published elsewhere.49 The thin film was prepared by treating the crystal with 20 μl of a 0.81 g L−1 (1 mM) dichloromethane solution of DEC+NO3−. Evaporation of dichloromethane left a thin-film coating of DEC+NO3− on the surface of the ATR crystal. The film thickness was determined to be ∼0.1 μm by ellipsometry (WVASE 32TM, J.A. Wollman Co., Inc., Lincoln, NE). The coated ATR probe was immersed into 100 mL of a stirred (200 rpm) aqueous sample and a spectrum (64 co-added scans) was recorded every minute for 15 min. At this time, the film, which now contained DEC+ClO4− in some cases as well as DEC+NO3−, was removed using dichloromethane and replaced with a fresh coating of DEC+NO3− for the next analysis. The relevant ion-exchange equilibrium is:44–46
| DEC+NO3−(film) + ClO4−(aq) ⇌ DEC+ClO4−(film) + NO3−(aq) |
In a typical analysis, the coated SiComp® probe was immersed in 100 mL of DI water for 10 min and a new single-beam background was collected. An appropriate volume of one of the fertilizer stock solutions was then added to make a solution for ATR-FTIR analysis that was either 100 mg L−1 (for fertilizers #2 and #4) or 1000 mg L−1 (for fertilizers #1, #3, #5, #6, and #7) in fertilizer concentration. The aforementioned 15 minute-by-minute spectra were then recorded. The set of spectra for fertilizer #2 (which is typical for samples that contained detectable perchlorate) is shown in Fig. 2. Each sample solution was analyzed in triplicate (i.e., using a fresh coating of DEC+NO3− for each of the three analyses).
 | ||
| Fig. 2 ATR-FTIR spectra of an aqueous solution of 100 mg L−1 of fertilizer sample #2 listed in Table 1 collected every minute for 15 min using the SiComp® probe coated with a thin film of the ion-exchange compound DEC+NO3−. | ||
Several of the fertilizer solutions gave a positive indication for the presence of perchlorate by the growth of the ν3 peak of perchlorate in the film at 1096 cm−1 as ion-exchange equilibration took place. We have established that the initial rate of growth of an absorbance peak associated with a particular anion is linearly related to the concentration of that anion in solution when exposed to an ATR probe coated with the appropriate ion-exchange film. For these fertilizer solutions, quantification of perchlorate was determined by the method of standard additions, using the initial rate of peak growth as the dependent variable.
First, a plot of the absorbance of the 1096 cm−1 peak vs. time was made, and the slope of the initial, apparently linear, portion (dA(1096 cm−1)/dt) was determined. This plot for sample #2 is shown in Fig. 3a. Note that, while the plot is apparently linear from 0 to about 15 min (the region over which the slope was determined), the overall approach to ion-exchange equilibrium is apparently exponential.
 | ||
| Fig. 3 (a) Plot of absorbance at 1096 cm−1vs. time for the ATR-FTIR spectra displayed in Fig. 2, along with those collected for the same sample from 16 to 60 min. The slope of the approximately linear initial portion of this graph, dA(1096 cm−1)/dt, was used as one of the data points in the standard addition graph below it. (b) ATR-FTIR standard addition graph for fertilizer sample #2 listed in Table 1. Using this graph, the absolute value of the x-axis intercept corresponds to the amount of perchlorate in the 100 mg L−1 fertilizer solution. After accounting for dilution, there is 340 ± 30 mg kg−1 perchlorate in this fertilizer. | ||
Next, each of the fertilizer solutions shown to contain perchlorate was analyzed several more times using this procedure, but with a known amount of a 0.11 g L−1 lithium perchlorate stock solution added to the sample. This procedure was repeated for at least four different concentrations of added perchlorate (three replicates each). Each perchlorate-spiked, standard-addition sample gave a dA(1096 cm−1)/dt value that was linearly related to the overall concentration of perchlorate in solution.
Finally, a plot of dA(1096 cm−1)/dt vs. the perchlorate concentration increment was made, and the absolute value of the x-axis intercept of a linear least-squares fit to the data was taken as the concentration of perchlorate in the unspiked fertilizer sample solution. The perchlorate concentration in the solid fertilizer sample was then calculated using this value and the known dilution factor. The plot of dA(1096cm−1)/dt vs. the perchlorate concentration increment for sample #2 is shown in Fig. 3b.
Raman spectroscopy
Extracts for perchlorate determination by Raman analysis were prepared using a protocol similar to that described by Williams et al.23 Briefly, for the solid fertilizers (i.e., samples #1-#5 of Table 1), about 5 g of the riffled material was extracted with 20–25 mL of DI water with an 18 MΩ cm resistivity. The mixture/solution was vortexed for 1 min and subsequently filtered through a Millipore Millex-HV 0.45 μm poly(vinylidene fluoride) (PVDF) filter. Note that perchlorate was not detected by Raman spectroscopy in sample #1 using these conditions. Later, after perchlorate was observed with the three other techniques, an additional extract of solid fertilizer sample #1 was prepared in a similar fashion, but using a higher solid-to-water ratio (i.e., 5 g in 5 mL). A 2 mL portion of this more concentrated extract of fertilizer sample #1 was treated (after filtration) with 1 g of activated alumina adsorbent (DD6 by Alcoa), in order to decrease the very high level of some of the fertilizer nutrients. The treatment with DD6 generally followed the procedure of Ellington and Evans,16 who have shown that DD6 significantly decreases (by competitive sorption) the concentration of ionic fertilizer nutrients in aqueous samples with no detectable loss of perchlorate when the nutrients are present at higher levels, as is the case here. Perchlorate was observed with Raman analysis in this more concentrated extract and was confirmed (both qualitatively and quantitatively) using a spike-and-recover procedure.The liquid fertilizers (samples #6 and #7 of Table 1) were analyzed for perchlorate directly after passing 5 mL aliquots through Millex-HV filters. The aliquots were taken from the original containers that where hand-shaken for at least 2 min.
Separate extracts for nitrate determination by Raman analysis were prepared in a similar fashion, but using a much lower fertilizer-to-water w/w ratio. In this case, for both solid and liquid fertilizers, about 0.14 g of fertilizer was dissolved in 25 mL of dionized water.
The Raman spectra were acquired with a Kaiser Optical Systems HoloProbe, using 785 nm laser excitation from a 300 mW SDL-8530 external-cavity-stabilized diode laser. This type of Raman instrument has been described fully elsewhere.23,50 Briefly, light from the laser was coupled to a remote probe head via a fiber optic cable. This laser light was then brought to focus with a series of lenses at approximately 7.5 cm beyond the end of the probe head assembly. A standard quartz cuvette, containing the aqueous fertilizer solution, was placed in the path of the beam such that the focus of the beam fell in the center of the solution. The power of the laser light at the solution was generally about 100 mW.
Raman scattered light from the solution was collected by the probe head along the same path as the excitation laser beam (i.e., 180° backscattering geometry), and was coupled to a separate fiber optic cable for delivery to the spectrograph. Inside the spectrograph, the Raman scattered light was focused through a 50 μm slit and directed through a volume holographic grating and focused onto the charge-coupled device (CCD) detector. This spectrograph permits acquisition of the entire Raman spectrum, with a useable Stokes Raman shift of about 3280 to 95 cm−1 in a single exposure, with about 5 cm−1 spectral resolution. The CCD detector uses a high quantum efficiency Princeton CCD-1024EHRB back-illuminated, deep depletion, near-infrared-optimized chip, which is thermoelectrically cooled to −65 °C. A Kaiser Optical Systems, Inc., HoloLab calibration accessory was used in determining system response for frequency and intensity calibration.51
For perchlorate determinations, Raman spectra were typically collected with an exposure time of 20 s with five accumulations co-added. Spectra were collected in this fashion for each of the seven fertilizer sample solutions, for numerous perchlorate standards in water, and for pure water. These settings resulted in a total analysis time of ∼7 min per spectrum. In a few cases, a higher exposure or more accumulations were employed to improve S/N for solutions containing lower perchlorate levels, thereby increasing the total analysis time to about 40 min. No solution exhibited significant fluorescence under any conditions.
For nitrate determinations, Raman spectra of extracts, standards, and water were collected in a similar manner. However, in this case, all spectra were collected with an exposure time of 15 s with three accumulations co-added. This resulted in a total analysis time of ∼3 min per spectrum.
All sample and standard solutions were analyzed in triplicate. The perchlorate concentrations in the hydroponic fertilizer solutions were determined for all spectra based on the fitted area of the symmetric ClO4 stretching peak (ν1),47 which occurs near Raman shift of 934 cm−1. (A linear calibration curve was constructed using perchlorate standards prepared in DI water. Details of the method of quantification can be found elsewhere.)23 Then, the perchlorate concentrations for the solid fertilizer samples were determined based on the solid-to-water ratio of the extracts, assuming 100% extraction efficiency. Finally, the mean perchlorate concentration of each solid sample was determined, along with the sample standard deviation (s); these values are reported in Table 1.
The nitrate concentrations in the fertilizer extracts, and in the original solid and liquid products, were determined in the same manner using the fitted area of the symmetric NO3 stretching peak (ν1),47 which occurs near Raman shift of 1047 cm−1. In Table 2, we present these results as ‘% nitrogen as nitrate,’ which is the weight percent of the fertilizer product that is nitrogen occurring as nitrate.
The following reagents were used as calibration standards: ammonium perchlorate (NH4ClO4) 99+%, Aldrich; sodium perchlorate (NaClO4) 99+%, Acros; sodium nitrate (NaNO3) 99+%, Fisher. Each was used as received without further purification.
Results and discussion
Unequivocal identification of perchlorate, particularly in complex aqueous samples, is a challenging analytical problem. For obvious reasons, many convenient and widely accepted techniques for definitive analysis of environmental contaminants (e.g., GC-MS) cannot be applied directly to inorganic ions occurring in aqueous media. The most common technique for perchlorate analysis is IC with suppressed conductivity detection.7,52 IC is widely available, exhibits a low LOD for perchlorate in relatively pure water (∼1 ppb), and can be equipped with autosamplers for unattended operation. However, identification is based solely on retention time matching since all ions are amenable to conductivity detection. Also, analysis by IC is greatly hindered when samples contain high total dissolved solids (TDS), as is the case with these hydroponic fertilizer solutions; TDS roughly equates to ionic strength. Both qualitative and quantitative analysis of perchlorate in fertilizers is compromised because the small perchlorate peak elutes on the tail of a very large peak due to the major fertilizer components. Samples generally must be diluted to bring the TDS down to a manageable level. This raises the w/w LOD (relative to that for perchlorate in pure water), typically by orders of magnitude. While detection limits will vary from fertilizer to fertilizer, previous results17,27,28 and the work here indicate a typical w/w LOD of about 30 mg kg−1. Quite often, a series of runs is necessary in order to find the level of dilution that yields acceptable results. This significantly increases the total analysis time. Also, high TDS samples rapidly degrade the IC column and foul the suppressor and detector.For qualitative chemical identifications to be considered definitive, a detection method more specific than retention-time matching is usually required. Historically, information-rich spectroscopic methods (e.g., MS, IR, and NMR) have been used to satisfy this need. Certain types of these methods have been coupled to chromatographic instruments and have become valuable and routine tools for analysis of multi-component samples (e.g., GC-MS, GC-IR, and LC-MS).
Unfortunately, none of these off-the-shelf coupled tools are directly amenable to analysis of perchlorate in fertilizer extracts. In some cases, the limitation is imposed by the separation technique. Fortunately, although fertilizer extracts are complex (i.e., containing very high levels of TDS), the total number of unique components is limited, and chromatographic separation is not required if the detection method has adequate selectivity. All of the spectroscopy-based approaches described here rely on this principle. However, the selectivity afforded by each technique differs, one from the other, in the physical property of perchlorate upon which the measurement depends. The qualitative and semi-quantitative agreement observed among these techniques provides a highly confident determination of perchlorate in the hydroponic fertilizers tested (see Table 1). It is useful to briefly discuss a few salient points of the application of each spectroscopic technique to hydroponic fertilizer analysis to illustrate how these techniques together contribute to these results.
cESI-MS analysis
The use of cESI-MS provides confident identification of low molecular weight ions by MS without prior analyte separation.10,53–55 The cESI-MS method is the most sensitive for detection of perchlorate in relatively pure water (LOD ∼0.3 ppb) among those described here. Perchlorate determination by cESI-MS involves the use of a quaternary ammonium ion that forms an ion pair with perchlorate that can be selectively extracted into dichloromethane, and has been described in detail elsewhere.10,53 The procedure allows the extraction of perchlorate from water, which is a problematic matrix for MS detection. This approach provides excellent selectivity, and is definitive for perchlorate identification. Unfortunately, cESI-MS is adversely affected by high TDS, roughly to the same degree as IC, based on difficulties in quantification. Other anions present in the aqueous sample (in this case, fertilizer nutrients) also form complexes with the quaternary ammonium base molecules and are extracted along with the perchlorate complex. High TDS affects the MS background signal and reduces the efficiency with which the electrospray apparatus produces perchlorate-complex ions. As with IC, the net effect is to raise the w/w LOD (relative to that in pure water), due to the need to dilute the sample. The selectivity, however, is not significantly affected by high TDS because of the unique mass spectrum of the perchlorate complex.As described in the Experimental section, samples that were subjected to cESI-MS analysis were obtained from two successive IC fractionation runs. The organic extracts of the final fractions (after addition of the complexing agent) were injected into the ESI-MS instrument and the mass spectra of the perchlorate complexes were obtained. The most sensitive and selective method of analysis involves single ion monitoring using m/z 380, which corresponds to the most abundant perchlorate complex anion: C10H21N(CH3)3Br(ClO4)−. An example of three successive 50 μL injections of the organic extract for fertilizer sample #1 is shown in Fig. 4.
 | ||
| Fig. 4 cESI-MS single ion monitoring (negative ion mode) spectrum for three successive 50 μL injections of a CH2Cl2 extract of the IC eluate fraction collected at 9.0–11.0 min, whose IC chromatogram is shown in Fig. 1(c), after complexation. The most abundant complex anion, C10H21N(CH3)3(Br)(ClO4)–, peak is observed at m/z 380. | ||
The experiments reported here are not useful for estimating LOD’s of cESI-MS for perchlorate directly in fertilizer extracts because isolated IC fractions, which were much lower in TDS, were used. Some of us have previously used cESI-MS for analysis of perchlorate in fertilizers without IC fractionation and have observed LOD’s of about 100 mg kg−1 or less.28 In the present work, the LOD’s are reduced by incorporating IC fractionation; however, the improvement is limited by the concomitant dilution. The LOD of cESI-MS with IC fractionation as described herein is expected for most samples to be at least as low as that of IC (i.e., ∼ 30 mg kg−1).
ATR-FTIR analysis
Attenuated total reflectance FTIR spectroscopy is directly applicable to determination of polyatomic ions such as perchlorate in water.48 However, using a commercially available 30-bounce silicon ATR probe, the perchlorate LOD of 230 ppm in DI water is rather high. The method described herein (and in more detail elsewhere)49 involves the use of a thin-film coating consisting of a water-insoluble organometallic ion-exchange compound that is selective for perchlorate on the ATR crystal. When the modified probe was immersed in a perchlorate-containing aqueous solution, nitrate ions in the thin film were replaced by perchlorate ions, and an infrared spectral peak corresponding to perchlorate (the asymmetric ClO4 stretching vibration, ν3, which is of T2-symmetry and occurs at 1096 cm−1)47 appeared and grew in intensity over time. The initial rate of growth of the absorbance of this peak, dA(1096 cm−1)/dt, is linearly correlated with the concentration of perchlorate in solution. Use of the extractant-coated probe leads to a lowering of the LOD from 230 ppm to 15 ppb in DI water.49Although a new thin-film coating is required for each analysis, a relatively high degree of reproducibility can be achieved. For example, when three replicate films were treated with a given standard aqueous solution of lithium perchlorate, the dA(1096 cm−1)/dt values typically varied by ±10% about the mean over the concentration range 15 ppb to 1 ppm. The high TDS content and high nitrate concentrations of the aqueous fertilizer solutions required the use of the method of standard additions, and the reproducibility of dA(1096 cm−1)/dt values for a given sample varied from ±50% relative error for small amounts of added perchlorate to ±14% relative error for large amounts of added perchlorate. In addition, the high TDS and high nitrate content of the fertilizer solutions had an adverse effect, in most cases, on the perchlorate LOD. For example, a control experiment showed the perchlorate LOD is 0.6 ppm in a 1.55 g L−1 aqueous solution of nitrate (added as sodium nitrate), whereas the perchlorate LOD is 0.015 ppm in DI water for a 15 min contact.
The ATR-FTIR results for perchlorate and nitrate are shown in Tables 1 and 2, respectively. Note in Table 2 that the seven hydroponic fertilizers were found to contain very different amounts of nitrate, with the weight percent nitrogen as nitrate varying from about 4.1% to 16.7% (i.e., weight percent nitrate 18% to 74%). Except for perchlorate and nitrate, the components of the fertilizers were not determined in this study. However, the manufacturers’ listed N-P-K grades, which are shown in Table 1, indicate that the fertilizers also contained various amounts of phosphate and potassium. Therefore, each fertilizer, when dissolved in water, resulted in a unique aqueous matrix.
Perchlorate was not detected by any technique in four of the seven hydroponic fertilizers. Extracts of these four samples were used to estimate the LOD for perchlorate in fertilizer solutions using the extractant-coated SiComp® probe. Specifically, we determined the amount of added perchlorate that was required to obtain an absorbance peak at 1096 cm−1 with height equal to three times the peak-to-peak noise level at ∼1100 cm−1 for 64 co-added scans taken after an equilibration time of 15 min. The concentration of perchlorate in the extract at that point (assuming it contained zero perchlorate prior to addition) was taken as the estimate of LOD for that extract. As expected, the LOD was somewhat different for each fertilizer solution because each one represents a different matrix of dissolved solids. Specifically, LOD estimates ranged from 200 to 700 ppb, with the lower LOD estimates generally being observed for fertilizers with lower levels of nitrate.
Raman analysis
Perchlorate is measured in the bulk aqueous fertilizer solutions using normal Raman spectroscopy without complexation or sorption onto a surface. This is convenient, but not sensitive. Approximately 20 ppm is the LOD for perchlorate in DI water for Raman analysis (using the conditions described above) when no post-run data manipulation is performed. This is based on S/N = 3, using the height of the 934 cm−1 peak of perchlorate relative to peak-to-peak noise in the baseline in the same region of the spectrum. Subtraction of a spectrum of DI water (i.e., an ideal blank) decreases the Raman LOD to about 10 ppm, which is still more than two orders of magnitude above that of the other techniques described here for DI water.Although not very sensitive, Raman is a useful tool in this application because it can easily tolerate very high levels of TDS with only limited impact on the perchlorate LOD. Raman essentially has no upper limit of detection; therefore, samples that are very concentrated in both analyte and in other matrix components can be analyzed if there is adequate resolution of matrix-component and analyte peaks. (It has been shown recently that the Raman spectral features of common fertilizer components do not directly interfere with the 934 cm−1 peak of perchlorate.)23 Indeed, the LOD for perchlorate in concentrated fertilizer extracts is only slightly higher than that for DI water. The small increase is due to the Raman scatter and luminescence (and the associated shot noise) from various matrix components, which make it somewhat more difficult to discern the Raman peak from perchlorate. While this increase in LOD varies depending on the type of fertilizer, it is typically only about a factor of two to five. Also, for fertilizer extracts, the benefits of water subtraction are not as great (compared to perchlorate standards in water) because DI water no longer constitutes an ideal blank. For extracts of these hydroponic fertilizers, which contain highly water soluble nutrients but exhibit no appreciable fluorescence at this excitation wavelength, the LOD is about 50 ppm. Using our typical solid-to-water ratio of 1/4, this corresponds to a LOD of about 200 mg kg−1 for a fertilizer on a solid weight basis. Note that we have successfully detected perchlorate using a solid-to-water ratio as high as 1/1 (i.e., for sample #1 using DD6 treatment of the extract), which sets the LOD at about 50 mg kg−1. Given the high TDS tolerance of Raman spectroscopy, the w/w LOD for perchlorate in a solid (or liquid) fertilizer is no more than about one order of magnitude higher than that of IC and the other techniques described here.
As shown in Fig. 5, direct Raman analysis also allows some other fertilizer components (e.g., nitrate, phosphate, sulfate, and urea) to be measured simultaneous, along with perchlorate. This capability, which is not present with methods that rely on selective complexation, is important in some situations. For example, the Raman spectrum can serve as a fertilizer brand signature because peak intensities from the various components are unique for a given formulation. This serves as a useful quality control feature when multiple products or multiple lots of a given product are analyzed comparatively.
 | ||
| Fig. 5 Raman spectrum of a 0.2 g mL−1 solution of fertilizer sample #2 listed in Table 1. The spectrum was recorded at ∼5 cm−1 resolution with an exposure time of 20 s with five accumulations co-added. Laser power at the sample was approximately 100 mW. | ||
In spite of the simultaneous presence of signal from multiple fertilizer components, Raman is adequately selective for perchlorate because Raman-active fertilizer nutrients (mostly inorganic ions) are relatively few, and each exhibits only one or two intense Raman bands that are well separated from those of the other components. On the other hand, direct Raman analysis without analyte separation is often not feasible for applications where numerous organic components are present, due to overlapping Raman bands and also to the presence of fluorescence. The Raman spectrometer described here relies on laser excitation at 785 nm, which avoids fluorescence from many organic compounds. In spite of this fact, we previously have observed significant fluorescence when analyzing for perchlorate in plant tissues and in a few fertilizer extracts.23 However, significant fluorescence was not observed with any of the fertilizer solutions described here.
Perchlorate to nitrate ratios
Three of the five solid fertilizers listed in Table 1 were found to have detectable levels of perchlorate. Although none of these fertilizers have known links to caliche, we wondered whether their perchlorate concentrations were consistent with those in this mined ore. We have investigated this by comparing the perchlorate-to-nitrate concentration ratio (w/w) of the extracts of these fertilizers to that determined from published data21 on the concentration of water-soluble saline components in mined Chilean caliche. The historical minimum and maximum ratios that we calculated, 3.7 × 10−4 and 9.0 × 10−2, respectively, were based on eight samplings of large tonnages of ore mined by nitrate companies in northern Chile over a period of about 50 years.21 Therefore, if the source of perchlorate in a solid fertilizer product originates solely from mined Chilean caliche, one would expect the perchlorate-to-nitrate concentration ratio in its aqueous extract to be within or near the range set by these limits. While this is clearly not a definitive test, it may be useful as a screening tool. For example, results from this test may influence a decision to investigate potential sources of perchlorate other than Chilean caliche.Note that some of us previously have used Raman spectroscopy to conduct a perchlorate-to-nitrate concentration ratio test for a different set of fertilizers that were found to contain perchlorate.23 Based on that test, we argued that it was not reasonable to attribute the source of perchlorate in those products solely to the presence of components derived from Chilean caliche, because, in many cases, the ratio significantly exceeded the upper limit set by the historical data. This earlier test was conducted by comparing directly the area of the Raman peak from perchlorate to that of nitrate in a single spectrum.23 Here we have used a similar approach using Raman data, but have quantified independently the levels of nitrate and perchlorate in the solid fertilizers based on calibration curves and using two separate sets of extracts—one for nitrate analysis and one for perchlorate analysis. While less convenient, this new approach is probably more accurate, particularly when nitrate and perchlorate levels are vastly different and when very high solid-to-water ratios are needed to detect perchlorate by Raman analysis.
In addition, in this study we determined perchlorate-to-nitrate concentration ratios (w/w) with ATR-FTIR spectrscopy. As with the Raman work, both nitrate and perchlorate levels were measured using calibration curves based on the appropriate standards (as described in the Experimental section). The nitrate analyses were conducted with the uncoated ATR crystal, whereas the perchlorate analyses were conducted with the film-coated ATR crystal. Note that we could have determined perchlorate-to-nitrate concentration ratios with IC as well, but this would have been less convenient than the vibrational spectroscopic measurements because different IC columns and different IC methods are needed to measure nitrate and perchlorate. Given the agreement between Raman and ATR-FTIR measurements of perchlorate-to-nitrate concentration ratios, these IC measurements were not pursued.
Table 2 shows the results of perchlorate-to-nitrate concentration ratio determinations for the perchlorate-containing samples listed in Table 1 and for Bulldog Soda (SQM North America Corporation). Bulldog Soda is sodium nitrate derived solely from mined Chilean caliche and is sold as a single-component fertilizer product (N-P-K grade 16-0-0). The evaluation of Bulldog Soda serves as an independent assessment of the perchlorate-to-nitrate concentration ratio test. If this test is useful, then the ratio for Bulldog Soda (acquired ∼January 2000) would fall between the minimum and maximum levels (3.7 × 10−4 and 9.0 × 10−2, respectively) determined from the mined nitrate ores. Indeed, our measurements showed that the ratio for Bulldog Soda was within the expected range. (Note that our sample of Bulldog Soda, and all of these hydroponic fertilizers, were acquired before SQM modified its process to reduce the perchlorate concentration in its products.)
The concentration of nitrate in the fertilizer products is presented in Table 2 as ‘% nitrate nitrogen’ so that our measurements can be compared to the concentration of nitrate as reported on the fertilizer product package. Note that our measurements, which reflect the nitrate level in small laboratory samples, are not to be taken as a challenge to the manufacturers’ reported levels, which are representative of much larger amounts of product. Instead, we have presented our results in this manner to demonstrate their generally good agreement with the manufacturers’ reported levels.
As shown in Tables 1 and 2, all of our hydroponic fertilizers list the majority, if not all, of their nitrogen content as nitrate; however, we do not know the original source of nitrate in these samples. Nonetheless, we note that all perchlorate-containing hydroponic products—three solid fertilizers—list potassium nitrate on the label as one of the chemicals from which the nutrients were derived. (As stated earlier, one industrial process for production of potassium nitrate involves Chilean caliche as a starting material.) Further, we note that the two solid fertilizers with no detectable perchlorate do not list potassium nitrate. However, there is no clear pattern here because potassium nitrate is listed for the 2 liquid fertilizers, neither of which contain detectable perchlorate.
For evaluating the perchlorate-to-nitrate concentration ratios, it is interesting to note that the range of this ratio for the historical mining data is quite large—the maximum level is about 243 times the minimum level. However, the range of this ratio for the products in Table 2, including Bulldog Soda, is considerably smaller—the maximum level is only about 15 times the minimum level. Additionally, it is interesting to note that all of the values for the fertilizers, including Bulldog Soda, are clustered near the lower level of the historical range for the mined ore. For these reasons, we argue that the perchlorate-to-nitrate concentration ratios are generally consistent with mined Chilean caliche even though the value for sample #1 falls just below the minimum level of the historical mining data. Further, we note that the ratios provide evidence of the resemblance of these samples to our sample of Bulldog Soda, all of which were likely manufactured near the same time. Based on these observations, we cannot eliminate mined Chilean caliche as the sole source of perchlorate in these three hydroponic products. In any event, it is important to point out that improvements in the refinement process for Chilean caliche are resulting in a substantial reduction of the amount of perchlorate in fertilizer products containing components derived from Chile saltpeter. However, it is not known how long after implementing the new process it will take before all old stocks of Chile saltpeter-derived materials (such as Bulldog Soda) are depleted and only new stocks, prepared by the modified process, find their way into commercial use. Also, it is important to recall that perchlorate-free synthetic sources of nitrogen overwhelmingly dominate the US fertilizer marketplace.
In conclusion, IC, cESI-MS, normal Raman spectroscopy, and FTIR spectroscopy with an extractant-coated ATR crystal have been used to determine perchlorate at relatively low levels (100–350 mg kg−1) in some hydroponic fertilizer products. Our results are reported with a high level of confidence based on generally good agreement among the techniques. These spectroscopic methods provide much more definitive qualitative results than those obtained by the conventional perchlorate identification method of IC retention time matching. The cESI-MS and the extractant-coated ATR-FTIR method also exhibit high inherent sensitivity for perchlorate, but the w/w LODs and the convenience of application are limited, to some degree, by high TDS. The sensitivity of Raman spectroscopy is lower, but it exhibits inherently high TDS tolerance, and multiple fertilizer components often can be measured simultaneously along with perchlorate. Raman, and also the uncoated ATR-FTIR probe, were useful in determining perchlorate-to-nitrate (w/w) ratios for the perchlorate-containing products. The techniques described here, when used in concert, offer a particularly powerful approach since all depend on a different property of perchlorate for identification.
An analytical approach such as ours, using multiple complementary spectroscopic techniques, may allow a better understanding of perchlorate in hydroponic fertilizers as they become more-and-more used worldwide. However, we caution that the products tested here may not be representative of those used in large scale hydroponic farming, but the matrixes appear to be representative of the macronutrients used in all hydroponics. Also, our results do not necessarily suggest a continuing or constant level of perchlorate, even in these tested products. Because fertilizer manufacturers vary the sources of their raw materials, it is likely that new lots of these same products will contain different levels of perchlorate, perhaps even be perchlorate-free. Finally, it is important to note that these hydroponic products are definitely not representative of those fertilizers used for production agriculture, which have recently been shown to be perchlorate-free.25 Indeed, at the present, we are not aware of any perchlorate-contaminated site that has been linked to hydroponic fertilizers or to any fertilizers whatsoever.
Acknowledgements
Fruitful discussions with J. J. Ellington and A.W. Garrison, and some technical assistance from D. E. Norton, J. Washington, and J. Evans, in Athens are appreciated. The work at Colorado State University was funded by NSF grant CTS-0085892.References
- J. J. J. Clark, in Perchlorate in the Environment, ed. E. T. Urbansky, Kluwer/Plenum, New York, 2000, ch. 3 Search PubMed.
- US EPA, Perchlorate environmental contamination: toxicological review and risk characterization based on emerging information. Second external review draft, NCEA-1-0503, US Environmental Protection Agency, Office of Research and Development, National Center for Environmental Assessment, Washington, DC, 2002 Search PubMed.
- E. T. Urbansky, Environ. Sci. Pollut. Res., 2002, 9, 187 Search PubMed.
- Fed. Regist., 1998, 63(40), p. 10274 Search PubMed.
- Fed. Regist., 1999, 64(180), p. 50555 Search PubMed.
- P. E. Jackson, M. Laikhtman and J. S. Rohrer, J. Chromatogr., A., 1999, 850, 131 CrossRef CAS.
- P. E. Jackson, S. Gokhale, T. Streib, J. S. Rohrer and C. A. Pohl, J. Chromatogr., A., 2000, 888, 151 CrossRef CAS.
- P. E. Jackson, S. Gokhale and J. S. Rohrer, in Perchlorate in the Environment, ed. E. T. Urbansky, Kluwer/Plenum, New York, 2000, ch. 5 Search PubMed.
- M. L. Magnuson, E. T. Urbansky and C. A. Kelty, Talanta, 2000, 52, 285 CrossRef CAS.
- M. L. Magnuson, E. T. Urbansky and C. A. Kelty, Anal. Chem., 2000, 72, 25 CrossRef CAS.
- E. T. Urbansky and M. R. Schock, J. Environ. Manage., 1999, 56, 79 Search PubMed.
- E. T. Urbansky, B. Gu, M. L. Magnuson, G. M. Brown and C. A. Kelty, J. Sci. Food Agric., 2000, 80, 1798 CrossRef CAS.
- R. Handy, D. A. Barnett, R. W. Purves, G. Horlick and R. Guevremont, J. Anal. At. Spectrom., 2000, 15, 907 RSC.
- B. Ells, D. A. Barnet, R. W. Purves and R. Guevremont, J. Environ. Monit., 2000, 2, 393 RSC.
- E. T. Urbansky, M. L. Magnuson, C. A. Kelty and S. K. Brown, Sci. Total Environ., 2000, 256, 227 CrossRef CAS.
- J. J. Ellington and J. J. Evans, J. Chromatogr., A, 2000, 898, 193 CrossRef CAS.
- J. J. Ellington, N. L. Wolfe, A. W. Garrison, J. J. Evans, J. K. Avants and Q. Teng, Environ. Sci. Technol., 2001, 3213 CrossRef CAS.
- E. T. Urbansky, Biorem. J., 1998, 2, 81 Search PubMed.
- A. A. Schilt, Perchloric Acid and Perchlorates, GFS Chemicals, Inc., Columbus, OH, 1979. Search PubMed.
- E. T. Urbansky, S. K. Brown, M. L. Magnuson and C. A. Kelty, Environ. Pollut., 2001, 112, 299 CrossRef CAS.
- G. E. Ericksen, Geology and Origin of the Chilean Nitrate Deposits, US Department of the Interior, Washington, DC, 1981 Search PubMed.
- G. E. Ericksen, Am. Sci., 1983, 71, 366 Search PubMed.
- T. L. Williams, R. B. Martin and T. W. Collette, Appl. Spectrosc., 2001, 55, 967 Search PubMed.
- A. R. Lauterbach, ACS 222nd National Meeting, Chicago, USA, 2001 Search PubMed.
- E. T. Urbansky, T. W. Collette, W. P. Robarge, W. L. Hall, J. M. Skillen and P. F. Kane, Survey of Fertilizers and Related Materials for Perchlorate (ClO4−), EPA/6000/R-01/049, 2001 Search PubMed.
- S. Susarla, T. W. Collette, A. W. Garrison, N. L. Wolfe and S. C. McCutcheon, Environ. Sci. Technol., 1999, 33, 3469 CrossRef CAS.
- S. Susarla, T. W. Collette, A. W. Garrison, N. L. Wolfe and S. C. McCutcheon, Environ. Sci. Technol., 2000, 34, 224 CrossRef CAS.
- E. T. Urbansky, M. L. Magnuson, C. A. Kelty, B. Gu and G. M. Brown, Environ. Sci. Technol., 2000, 34, 4452 CrossRef CAS.
- W. P. Robarge, M. Duffera and G. Ramirez, ACS 220th National Meeting, Washington, DC, USA, 2000 Search PubMed.
- H. A. P. Ltd, Hydroponics as an Agricultural Production System’, 01/141, Rural Industries Research and Development Corporation, Sydney, NSW, Australia, 2001 Search PubMed.
- T. L. Giblin, D. C. Herman and W. T. Frankenberger, J. Environ. Qual., 2000, 29, 1057 Search PubMed.
- T. L. Giblin, D. C. Herman and W. T. Frankenberger, Jr, in Perchlorate in the Environment, ed. E. T. Urbansky, Kluwer/Plenum, New York, 2000, ch. 19 Search PubMed.
- J. D. Coates, U. Michaelidou, R. A. Bruce, S. M. O′Connor, J. N. Crespi and L. A. Achenbach, Appl. Environ. Microbiol., 1999, 65, 5234 CAS.
- J. D. Coates, U. Michaelidou, S. M. O′Connor, R. A. Bruce and L. A. Achenbach, in Perchlorate in the Environment, ed. E. T. Urbansky, Kluwer/Plenum, New York, 2000, ch. 24 Search PubMed.
- B. E. Logan, Biorem. J., 1998, 2, 69 Search PubMed.
- V. A. Nzengung, C. Wang and G. Harvey, Environ. Sci. Technol., 1999, 33, 1470 CrossRef CAS.
- J. Mason, Commercial Hydroponics, Kangaroo Press, Kenthurst, Australia, 1990 Search PubMed.
- B. Van Aken and J. L. Schnoor, Environ. Sci. Technol., 2002, 36, 2783 CrossRef CAS.
- J. B. Jones, Jr, Hydroponics—A Practical Guide for the Soilless Grower, St. Lucie Press, Boca Raton, 1997 Search PubMed.
- H. M. Resh, Hydroponic Food Production—A Definitive Guidebook of Soilless Food-Growing Methods, Woodbridge Press Publishing Company, Santa Barbara, 1995 Search PubMed.
- H. Soffer, in Hydroponics Worldwide: State of the Art in Soiless Crop Production, ed. A. J. Savage, International Center for Special Studies, Inc., Honolulu, 1985, p. 123 Search PubMed.
- J. F. Clark, D. L. Clark, G. D. Whitener, N. C. Schroeder and S. H. Strauss, Environ. Sci. Technol., 1996, 30, 3124 CrossRef CAS.
- B. J. Clapsaddle, J. F. Clark, D. L. Clark, K. M. Gansle, A. E. Gash, C. K. Chambliss, M. A. Odom, S. M. Miller, O. P. Anderson, R. P. Hughes and S. H. Strauss, Inorg. Chem., in preparation Search PubMed.
- C. K. Chambliss, M. A. Odom, C. M. L. Morales, C. R. Martin and S. H. Strauss, Anal. Chem., 1998, 70, 757 CrossRef CAS.
- C. K. Chambliss, M. A. Odom, C. R. Martin, B. A. Moyer and S. H. Strauss, Inorg. Chem. Commun., 1998, 1, 435 CrossRef CAS.
- C. K. Chambliss, C. R. Martin, S. H. Strauss and B. A. Moyer, Solvent Extr. Ion Exch., 1999, 177, 553 Search PubMed.
- R. A. Nyquist, C. L. Putzig and M. A. Leugers, The Handbook of Infrared and Raman Spectra of Inorganic Compounds and Organic Salts, Academic Press, San Diego, 1997 Search PubMed.
- R. N. Wilhite and R. F. Ellis, Appl. Spectrosc., 1963, 17, 168 Search PubMed.
- S. H. Strauss, M. A. Odom, G. N. Hebert and B. J. Clapsaddle, J. Am. Water Works Assoc., 2002, 94, 109 Search PubMed.
- N. Everall, H. Owen and J. Slater, Appl. Spectrosc., 1995, 49, 610 Search PubMed.
- J. M. Tedesco and K. L. Davis, Proc. SPIE-Int. Soc. Opt. Eng., 1998, 200 Search PubMed.
- T. W. Collette, W. P. Robarge and E. T. Urbansky, Ion Chromatographic Determination of Perchlorate Ion: Analysis of Fertilizers and Related Materials, EPA/600/R-01/026, US Environmental Protection Agency, Washington, DC, 2001 Search PubMed.
- E. T. Urbansky, M. L. Magnuson, D. Freeman and C. Jelks, J. Anal. At. Spectrom., 1999, 14, 1861 RSC.
- M. L. Magnuson, C. A. Kelty and R. Cantú, J. Am. Soc. Mass Spectrom., 2001, 12, 1085 CrossRef CAS.
- M. L. Magnuson and C. A. Kelty, Anal. Chem., 2000, 72, 2308 CrossRef CAS.
Footnote |
| † This paper has been reviewed in accordance with the US Environmental Protection Agency’s peer and administrative review policies and approved for publication. Mention of trade names or commercial products does not constitute endorsement or recommendation for use by the US EPA. This paper is the work of US government employees engaged in their official duties and is therefore exempt from US copyright protection pursuant to 17 USC 105. © US Government 2003 |
| This journal is © The Royal Society of Chemistry 2003 |