Luminescence of extended and folded N,N′-diarylureas
Frederick D.
Lewis
*,
Todd L.
Kurth
and
Weizhong
Liu
Department of Chemistry, Northwestern University, Evanston, IL 60208-3113, USA. E-mail: lewis@chem.northwestern.edu
First published on 2nd January 2002
Abstract
The relationship between molecular structure and luminescence of a series of secondary and tertiary N,N′-diarylureas (aryl = phenyl, 2-naphthyl, 2-anthryl, and pyren-1-yl) has been investigated at 77 K and at room temperature in the glass-forming solvent methyltetrahydrofuran. The secondary diarylureas possess planar extended structures, whereas the tertiary diarylureas possess folded structures in which the aryl groups are face-to-face. The secondary and tertiary diphenylureas display broad, weak fluorescence attributed to an n,π* singlet state at low temperature and are non-fluorescent at room temperature. The other secondary diarylureas are strongly fluorescent both at 77 K and at room temperature. Their fluorescence resembles that of the parent arene and is assigned to a lowest π,π* singlet state. The fluorescence of the other tertiary diarylureas is similar to that of the secondary diarylureas at 77 K, but is broad and red-shifted in fluid solution, as a consequence of intramolecular excimer formation. The properties of these novel intramolecular excimers are compared with those of 1,3-diarylalkanes and paracyclophanes.
Introduction
The formation of fluorescent intramolecular excimers has been reported for a number of 1,n-diarylalkanes.1,2 In the initial study of intramolecular excimer formation in 1,n-diphenylalkanes by Hirayama,3 excimer fluorescence was observed when n = 3, but not for longer or shorter linkers. Formation of fluorescent intramolecular excimers has subsequently been observed for both longer and shorter linkers connecting polynuclear aromatic hydrocarbons including naphthalene,4 anthracene,2,5,6 and pyrene.7,8 However, the trimethylene linker is optimal for the formation of a strain-free excimer possessing a face-to-face or “sandwich” geometry. The ground state population of the sandwich geometry is generally quite small for n-alkyl chains due to the unfavorable energetics of chain folding. As a consequence, only monomer fluorescence is observed in highly viscous media that inhibit chain folding.Significant populations of folded ground state dimer geometries can be obtained by means of linker design. For example, the racemic isomer of 2,4-dipyren-2-ylpentane exists as a ca. 4 ∶ 1 mixture of folded (gauche,gauche) and extended (trans,trans) conformers at room temperature.9 Bis(pyrenylcarboxy)alkanes10 and polyether-linked bis(hemicyanines)11 also adopt folded dimer geometries in nonpolar solvents due to the poor solvation of their polar functional groups. Excimer fluorescence has also been observed for 1,8-pyrenylnaphthalenes12 and for a large number of cyclophanes which possess arene-dimer ground state geometries.5,13,14 The photophysical behavior of the latter excimers is strongly correlated with the cyclophane geometry.
We report here the results of our investigation of the photophysical properties of the disecondary N,N′-diarylureas Ia–IIIa and the ditertiary N,N′-dimethyl-N,N′-diarylureas Ib–IVb (Scheme 1). Disecondary diarylureas are known to adopt extended structures both in solution and in the solid state.15,16 In contrast, N,N′-diethyl-N,N′-diphenylurea adopts a folded structure in which the phenyl groups are face-to-face.17 More recently, Yamaguchi et al.18 reported the structures of a three-layered naphthalene derivative with two tertiary urea linkers and a five-layered phenyl derivative with four tertiary urea linkers. Both of these molecules adopt π-stacked structures both in solution as well as in the solid state. The distinctive structures of the secondary and tertiary diarylureas have been exploited both in the control of molecular architecture and in molecular assembly.19,20 Curiously, the photophysical properties of these structurally unique molecules have not been investigated.
 | ||
| Scheme 1 | ||
Results
Preparation, conformation, and electronic absorption spectra
N,N′-Diphenylurea (Ia) and its N,N′-dimethyl derivative (Ib) are commercially available. The secondary diarylureas of naphthalene and anthracene (IIa and IIIa) are conveniently prepared from the reaction of the appropriate aminoarenes with triphosgene. Conversion of IIa and IIIa into their N,N′-dimethyl derivatives IIb and IIIb was accomplished by direct alkylation using sodium hydride and iodomethane. The tertiary monopyrenylurea IVa was prepared via the sequential addition of 1-aminopyrene and dimethylamine to triphosgene, followed by N-methylation. The cyclic diphenylurea Ic was prepared via the reaction of 1,2-dianilinoethane with triphosgene. The 1H NMR spectra of IIa and IIIa and IIb–IVb in CDCl3 solution are consistent with cis,cis and trans,trans structures, respectively, similar to those reported for Ia and Ib (Scheme 1).18,19 In all cases the NMR spectra consist of a single set of resonances at room temperature.The ground state structures of the diarylureas were investigated using AM1 calculations. The secondary diarylureas all adopt planar, extended structures. The minimized energies of the three rotamers of IIa (Scheme 2) are similar. The tertiary diarylureas adopt folded structures. The minimized structures of Ib and of the syn and anti rotamers of IIb are shown in Fig. 1. The calculated (gas phase) energy of the syn rotamer is lower than that of the anti rotamer by 2.2 kcal mol−1. The shortest distance between arene carbons is 2.98 Å for Ib and 2.99 Å for the syn rotamer of IIb and the dihedral angles between the two arenes in Ib and IIb are 33° and 35°, respectively. The calculated energy difference between extended cis,cis and folded trans,trans structures are −7 and −2 kcal mol−1 for Ia and IIa and 4 and 5 kcal mol−1 for Ib and IIb, respectively. In all cases the energies of the trans,cis rotamers were higher than those of the cis,cis rotamers of the disecondary ureas and the trans,trans rotamers of the ditertiary ureas.
 | ||
| Scheme 2 | ||
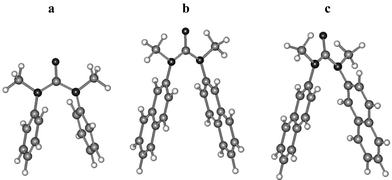 | ||
| Fig. 1 AM1-optimized geometries for (a) Ib, (b) anti-IIb, and (c) syn-IIb. | ||
The absorption spectra for the secondary and tertiary diarylureas at room temperature in methyltetrahydrofuran (MTHF) solution are shown in Figs. 2–5. Absorption by solvent distorts the spectra below 230 nm. The absorption maxima and molar absorbances for the resolved long-wavelength bands are reported in Table 1 along with data for the parent arenes.21 The band structures of the secondary diarylureas Ia–IIIa and the tertiary monoarylurea IVa are similar to those of the parent arenes. However, the long-wavelength absorption maxima (Band 1) are red-shifted compared to those of the parent arenes. The absorption bands of the tertiary ureas Ib–IIIb are broadened and blue-shifted by 1400 ± 200 cm−1 relative to those of the secondary ureas. The Band 1 molar absorbances are significantly larger for both the secondary and tertiary ureas than for the parent hydrocarbons, except in the case of IIIa and IIIb which have absorbances slightly smaller than that of anthracene (Table 1).
| λ/nm (log ε) | ||
|---|---|---|
| Band 1 | Band 2 | |
| a Data for arylureas in methyltetrahydrofuran solution (this study). Data for arenes in alkane solution from ref. 21. Reported wavelengths are for the first resolved vibronic band, when resolved, or for the band maximum. sh = shoulder. | ||
| Benzene | 254 (2.40) | |
| Ia | 256 (4.66) | |
| Ib | 246 (4.05) | |
| Ic | 265 (4.33) | |
| Naphthalene | 311 (2.43) | 286 (3.75) |
| IIa | 338 (3.93) | 298 (4.82) |
| IIb | 313 (sh) (3.48) | 290 (4.06) |
| Anthracene | 375 (3.93) | 252 (5.34) |
| IIIa | 402 (3.85) | 277 (4.86) |
| IIIb | 382 (3.81) | 250 (5.06) |
| Pyrene | 372 (2.71) | 334 (4.74) |
| IVa | 380 (sh) (3.07) | 348 (4.45) |
| IVb | 380 (sh) (3.60) | 334 (4.55) |
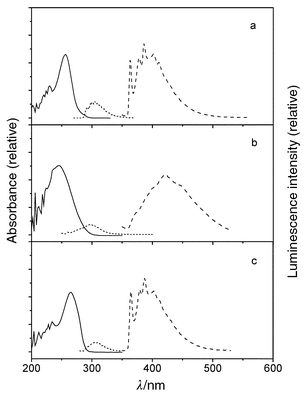 | ||
| Fig. 2 Room temperature UV (solid line) and 77 K fluorescence (short dash) and phosphorescence (dash) spectra for (a) Ia; (b) Ib; (c) Ic. | ||
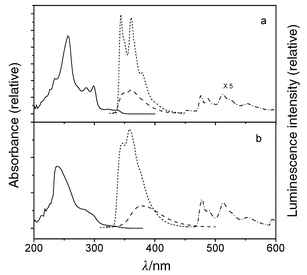 | ||
| Fig. 3 UV (298 K: solid), fluorescence (77 K: short dash, 298 K: dash), and phosphorescence (77 K: dash–dot) spectra for (a) IIa and (b) IIb. | ||
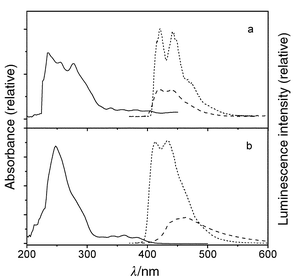 | ||
| Fig. 4 UV (298 K: solid) and fluorescence (77 K: short dash, 298 K: dash) spectra for (a) IIIa and (b) IIIb. | ||
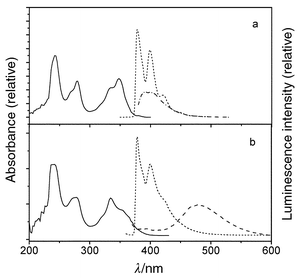 | ||
| Fig. 5 UV (298 K: solid) and fluorescence (77 K: dash, 298 K: dash–dot) spectra for (a) IVa and (b) IVb. | ||
| T/K | λ f/nm | Φ f | τ s/ns | 107kf/s−1 | |
|---|---|---|---|---|---|
| a Data for arylureas in MTHF solution. Data for arenes in alkane solution from ref. 21. Fluorescence wavelengths are for the first resolved band, when available, or for band maxima. b Dual exponential decay, minor component in parentheses. | |||||
| Benzene | 298 | 277 | 0.07 | 29 | 0.24 |
| Ia | 77 | 292 | 0.07 | 5.7 | 1.3 |
| Ib | 77 | 310 | 0.003 | 3.7 | 0.08 |
| Ic | 77 | 310 | 0.05 | 2.7 | 1.8 |
| Naphthalene | 298 | 322 | 0.27 | 96 | 0.28 |
| IIa | 77 | 345 | 0.91 | 15.5 (6.9)b | 13.0 |
| 298 | 350 | 0.24 | 8.0 | 3.0 | |
| IIb | 77 | 348 | 0.50 | 5.4 | 9.3 |
| 298 | 377 | 0.015 | 0.43 | 3.5 | |
| Anthracene | 298 | 400 | 0.32 | 4.9 | 6.5 |
| IIIa | 77 | 420 | 0.86 | 17.2 | 5.0 |
| 298 | 420 | 0.36 | 16.9 | 2.1 | |
| IIIb | 77 | 415 | 1.0 | 14.1 | 7.1 |
| 298 | 460 | 0.037 | 0.43 | 0.86 | |
| Pyrene | 298 | 385 | 0.32 | 450 | 0.07 |
| IVa | 77 | 380 | 0.73 | 31.0 | 2.3 |
| 298 | 390 | 0.41 | 11.7 | 3.5 | |
| IVb | 77 | 400 | 0.50 | 10.6 (38.3)b | 1.3 |
| 298 | 480 | 0.075 | 22.6 | 0.33 | |
The secondary diarylureas IIa and IIIa and the monoarylurea IVa all display intense fluorescence (Φf ≥ 0.73) and weak phosphorescence (Φp ≤ 0.05) at 77 K (Figs. 3–5). Upon warming to room temperature, broadening of the fluorescence accompanied by a small red-shift in the fluorescence maxima, and a decrease in Φf are observed. The 77 K fluorescence spectra of the tertiary diarylureas IIb–IVb are somewhat broader than those of IIa–IVa, but their maxima and decay times are similar. In the case of IIb, moderately intense, structured phosphorescence is also observed at 77 K. Upon warming to Tg the fluorescence spectra of IIb–IVb undergo large red-shifts and a decrease in intensity. Further warming to room temperature results in continuous decreases in Φf but no further shift in the fluorescence maxima.
The luminescence decays are best fitted by single exponential decay functions, except in the case of IIa and IVb at 77 K, for which dual exponential decay is observed. The fluorescence decay times of both the secondary and tertiary ureas are shorter than those of the parent arenes except in the case of IIIa and IIIb which have longer decay times than anthracene at 77 K (Table 2). The tertiary ureas IIb and IIIb have significantly shorter decay times at room temperature than at 77 K. The room temperature decay time for IVb is in between the two decay components at 77 K. The phosphorescence decay times of the phenyl and naphthylureas are similar to those of the parent arenes (Table 3).
| λ p/nmb | Φ p | τ t/s | |
|---|---|---|---|
| a Data for arylureas in MTHF glasses at 77 K. Data for arenes in hydrocarbon glasses at 77 K from ref. 22. b Phosphorescence wavelengths are for the 0,0 band. | |||
| Benzene | 339 | 0.15 | 4.5 |
| Ia | 360 | 0.22 | 2.0 |
| Ib | 360 | 0.008 | |
| Ic | 360 | 0.18 | 2.1 |
| Naphthalene | 469 | 0.033 | 2.3 |
| IIa | 475 | 0.05 | 2.2 |
| IIb | 475 | 0.17 | 1.6 |
| Urea | State | λ calcd/nm | 105fa | Character |
|---|---|---|---|---|
| a Calculated oscillator strength. | ||||
| Ia | S1 | 323 | 0 | n,π* |
| S2 | 277 | 148 | π,π* | |
| S3 | 277 | 129 | π,π* | |
| S4 | 255 | 6750 | π,π* | |
| Ib | S1 | 314 | 0 | n,π* |
| S2 | 278 | 10 | π,π* | |
| S3 | 284 | 137 | π,π* | |
| S4 | 242 | 1680 | π,π* | |
| IIa | S1 | 322 | 0 | π,π* |
| S2 | 322 | 65 | π,π* | |
| S3 | 311 | 14 | n,π* | |
| S4 | 283 | 3620 | π,π* | |
| IIb | S1 | 326 | 35 | π,π* |
| S2 | 308 | 3 | n,π* | |
| S3 | 287 | 112 | π,π* | |
| S4 | 287 | 2330 | π,π* | |
| IIIb | S1 | 363 | 283 | π,π* |
| S2 | 357 | 78 | π,π* | |
| S3 | 337 | 3350 | π,π* | |
| S4 | 313 | 468 | π,π* | |
| IVb | S1 | 399 | 513 | π,π* |
| S2 | 385 | 136 | π,π* | |
| S3 | 368 | 213 | π,π* | |
| S4 | 332 | 5390 | π,π* | |
Discussion
Ground state conformation
N,N-Diphenylurea (Ia) and several of its ring-substituted derivatives adopt extended planar cis,cis conformations both in the solid state and in solution.15,16 AM1 calculations for IIa indicate that its cis,cis conformation is also significantly more stable than either its trans,trans or cis,trans conformations. We have determined the crystal structure of the 1-naphthyl analog of IIa and find that it also exists in a planar cis,cis conformation.24 The 1H NMR spectra of IIa and IIIa are also consistent with cis,cis conformations in solution, the naphthalene or anthracene H-1 proton chemical shifts reflecting deshielding by the adjacent carbonyl group. AM1 calculations indicate that the energies of the three N-aryl rotamers in which H-1 is either syn or anti to the carbonyl oxygen (Scheme 2) have similar energies. The observation of time-averaged 1H NMR spectra for IIa and IIIa suggests that the three N-aryl rotamers interconvert rapidly at room temperature.N,N-Dimethyl-N,N′-diphenylurea (Ib) and several of its ring-substituted derivatives adopt folded trans,trans conformations with face-to-face aryl groups both in the solid state and in solution.17,18 The AM1-minimized structure of Ib (Fig. 1) is similar to that of the crystal. The crystal structure of the 1-naphthyl analog of IIb also exhibits a face-to-face geometry.24 AM1 calculations for IIb indicate that its trans,trans conformation (Fig. 1) is significantly more stable than either its cis,cis or cis,trans conformation. The calculated dihedral angles between the two N-aryl groups in Ib and the syn rotamer of IIb are 33° and 35°, respectively. Thus, the distances between the aromatic carbons directly attached to nitrogen are shorter (ca. 3.0 Å) than those for other pairs of aromatic carbons.
The 1H NMR spectra of IIb, IIIb, and IVb display substantial up-field shifts of their aryl protons when compared to their secondary analogs, indicative of folded structures in solution. The ditertiary ureas IIb, IIIb, and IVb can exist in either fully (syn) or partially (anti) overlapping N-aryl rotamers (Fig. 1b, c). In the case of IIb, the calculated energy (gas phase) of the syn rotamer is lower than that of the anti rotamer (Fig. 1) by 2.2 kcal mol−1. As is the case for the disecondary ureas, time-averaged NMR spectra are observed for IIb, IIIb, and IVb at room temperature.
The face-to-face geometry of the tertiary diarylureas is similar to that of 1,8-diarylnaphthalenes, which have been termed “open-chain (cyclo)phanes” or “protophanes.”25 The structures of the 1,8-dipyrenylnaphthalenes Va–Vc (Scheme 1) have been reported by Wahl et al.12 The distance between pyrene carbons attached to the naphthalene rings (2.94 Å) is similar to that for the tertiary diarylureas (3.0 Å). However, the pyrene rings of Va–Vc are more nearly co-planar than those of IVb and both the naphthalene and pyrene rings are distorted from planarity. These distortions are similar to those for [3.3]paracyclophanes and less pronounced than those observed for [2.2]paracyclophanes.25 The less rigid nature of the urea linkage results in larger arene–arene distances and dihedral angles.
Electronic absorption spectra
Absorption spectral data for lowest energy band of the diphenylureas and the two lowest energy bands for the other diarylureas are summarized in Table 1 along with data for the parent arenes. The absorption spectra of the secondary diarylureas are similar to those of the parent arenes, but the maxima are shifted to longer wavelength (Figs. 2–5). The spectra of the tertiary diarylureas are broader than those of the parent arenes, however, their maxima are at similar wavelengths. A marked increase in intensity, relative to that of the parent arene, is also observed for the absorption bands of both the secondary and tertiary diphenyl-, dinaphthyl-, and dipyrenylureas, but not the dianthracenylureas.The observed spectral bands of the diarylureas can be assigned, on the basis of ZINDO calculations, to π,π* transitions with varying degrees of configuration interaction (Table 4). Configuration interaction is responsible for the increased intensity of Band 1 for the urea derivatives of benzene, naphthalene, and pyrene, which have weak 1Lb bands, and for the decreased intensity for Band 1 of the urea derivatives of anthracene, which has a more intense 1La band.21 A forbidden n,π* transition is calculated to lie below the lowest energy π,π* transition of the diphenylureas Ia and Ib (Fig. 6), but above the lowest π,π* transitions of the other diarylureas. The red-shift of the secondary diarylureas' bands relative to the parent arenes may reflect conjugation of the co-planar arene and urea orbitals, as seen for the π and π* orbitals of Ia in Fig. 6a. No such conjugation is possible for the orthogonal arene and urea orbitals in the tertiary ureas.
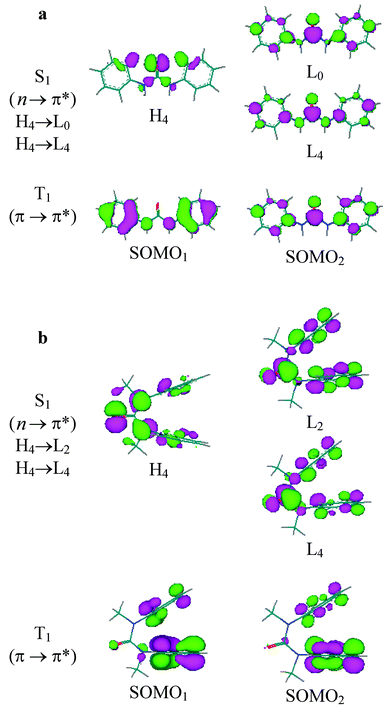 | ||
| Fig. 6 Nodal properties of orbitals for major components of S1 and T1 states for (a) Ia and (b) Ib. | ||
Broadening of the tertiary diarylurea absorption bands and skewing of the band shape toward shorter wavelength may reflect excitonic interactions between the π-stacked arene rings.26,27 These interactions are expected to be much weaker than those in [2.2]- or [3.3]paracyclophanes, in which both the arene–arene distances and arene–arene dihedral angles are smaller than those of the tertiary diarylureas.25 Based on their geometries, the 1,8-diarylnaphthalenes would be expected to display arene–arene electronic interactions stronger than those in the diarylureas. Unfortunately, detailed absorption spectral data for the 1,8-dipyrenylnaphthalenes Va–Vc have not been reported.12
Fluorescence of secondary diarylureas
Weak, structureless fluorescence is observed for the secondary diphenylurea Ia at 77 K (Fig. 2a). The fluorescence rate constant (kf) is larger than that of benzene, in accord with the greater intensity of Band 1 for Iavs. benzene (Table 1). The fluorescence intensity decreases markedly in fluid solution. The very weak fluorescence intensity in fluid solution (Φf < 10−3) no doubt accounts for the absence of prior reports of its fluorescence properties. Moderately intense phosphorescence is observed for Ia at 77 K (Fig. 2a), suggesting that efficient intersystem crossing may be responsible for the low fluorescence quantum yield. This behavior is consistent with the presence of a lowest energy n,π* singlet state, as indicated by the ZINDO calculations (Table 4).The secondary diarylureas IIa and IIIa display intense, structured fluorescence at 77 K in an MTHF glass (Figs. 3a, 4a). The spectra are broader but otherwise similar in appearance and Stokes shift to those of the parent arenes. The value of kf for IIa is much larger than that of naphthalene, whereas that for IIIa is slightly smaller than that of anthracene, in accord with the effects of urea substituents upon Band 1 intensities (Table 1). Upon warming to room temperature the spectra broaden, but only minor changes in the fluorescence maxima and lifetimes are observed (Table 2). This behavior is consistent with fluorescence from a planar lowest π,π* singlet state both at 77 K and room temperature. The calculated energy of the n,π* state of IIa is higher than that of the lowest π,π* state (Table 4). The fluorescence behavior of the tertiary monopyrenylurea IVa is similar to that of the secondary diarylureas IIa and IIIa, in accord with the absence of strong interactions between the aryl groups of the secondary diarylureas.
The fluorescence properties of the secondary ureas IIa and IIIa differ markedly from those of the N-arylbenzamides which display weak, highly Stokes shifted fluorescence in fluid solution.28,29 This fluorescence is attributed to a TICT state formed upon twisting about the amide N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. No TICT fluorescence is observed for the secondary diarylureas. The small thermochromic shifts for IIa and IIIa indicate that the geometry of the relaxed secondary diarylurea singlet state is similar to that of the planar ground state.
O bond. No TICT fluorescence is observed for the secondary diarylureas. The small thermochromic shifts for IIa and IIIa indicate that the geometry of the relaxed secondary diarylurea singlet state is similar to that of the planar ground state.
Fluorescence of tertiary diarylureas
The fluorescence of the tertiary diphenylurea Ib is broad and weak at 77 K (Fig. 2b) and is weaker still in fluid solution (Φf < 10−4). This is consistent with the presence of a lowest n,π* singlet state for Ib as well as Ia (Table 4). Both the fluorescence and phosphorescence quantum yields for Ib at 77 K are substantially smaller than those of Ia. The luminescence parameters of the cyclic urea Ic (Fig. 1c and Tables 2, 3) are more similar to those of Ia than of Ib. The phenyl groups in Ic are constrained to a cis,cis geometry. Thus, its excited state behavior appears to be determined by the urea conformation (trans,transvs. cis,cis) rather than by N-alkylation.The tertiary diarylureas IIb–IVb are strongly fluorescent at 77 K (Figs. 3b–5b). The fluorescence spectra at 77 K are somewhat broader than those for IIa–IVa, however, the fluorescence maxima, decay times, and fluorescence rate constants are similar for the secondary and tertiary ureas (Table 2). The fluorescence spectra of IIb–IVb are structureless and strongly red-shifted in fluid solution, indicative of intramolecular excimer formation. The observation of excimer fluorescence in fluid solution but not in the 77 K glass indicates that a large amplitude single state geometry change is necessary for excimer formation. In this respect, the behavior of the diarylureas is similar to that of the 1,3-diarylpropanes VIa–VId (Scheme 1), which also display excimer fluorescence in fluid solution, but not in the solid state.5 However, the diarylpropanes display temperature-dependent mixtures of monomer and excimer fluorescence in solution,4 whereas the tertiary diarylureas display only excimer fluorescence at all temperatures in fluid solution. This difference can be attributed to the preference of extended ground state conformers for the diarylpropanes vs. folded conformations for the diarylureas. Paracyclophanes with parallel arene rings separated by < 3.8 Å display excimer fluorescence in glasses at 77 K as well as in fluid solution.5,13,14
The monomer–excimer frequency shifts that are observed upon melting glasses containing IIb–IVb are reported in Table 5 along with the monomer–excimer frequency shifts for the diarylpropanes VIa–VId and the syn rotamer of 1,8-dipyren-1-ylnaphthalene Va.3,4,6,8 The frequency shifts for Va and VId are larger than that for IVb, plausibly reflecting more effective pyrene–pyrene π-stacking. Formation of a sandwich-type excimer with a ca. 3 Å π-stacking distance can occur readily for Va and VId but would require distortion of the urea bond angles in IVb.
| Arene | ArNMeCONMeAra | Ar(CH2)3Arb | 1,8-Naphthylc |
|---|---|---|---|
| Δνfl/cm−1 | Δνfl/cm−1 | Δνfl/cm−1 | |
| a Difference between the fluorescence maxima at 77 K and room temperature. b Difference between the monomer and excimer band maxima at room temperature. Data for VIa–VIb from refs. 3–8. c Difference between the band maxima of pyren-1-ylnaphthalene and 1,8-dipyrenylnaphthalene. Data from ref. 12. | |||
| Phenyl | VIa: 5500 | ||
| 2-Naphthyl | IIb: 2200 | VIb: 5100 | |
| 2-Anthryl | IIIb: 2900 | VIc: 5400 | |
| Pyren-1-yl | IVb: 4200 | VId: 5600 | Va: 5800 |
The diarylurea IVb can exist as a mixture of syn and antiN-aryl rotamers in solution. The syn rotamer of the rigid dipyrenylnaphthalene Va displays a much larger excimer shift than the anti rotamer Vb, presumably due to more extensive orbital overlap.12 However, investigation of excimer formation in three isomeric 1,3-dipyrenylpropenes with different pyrenyl ring substitution patterns showed that a partially-overlapping excimer is slightly more stable than the fully-overlapping excimer, plausibly due to reduced electronic repulsion.8 The urea linker is sufficiently flexible to allow similar π-overlap for the syn and antiN-aryl rotamers of the tertiary diarylureas. The observation of single exponential decay for IVb and the other tertiary diarylureas in solution indicates either that the two rotamers have similar fluorescence spectra and decay times, or that emission occurs from only one rotamer.
The monomer–excimer frequency shifts for the tertiary diarylureas IIb and IIIb are smaller than that for IVb (Table 5). This difference may reflect less extensive π-overlap for the smaller arenes. No such decrease in frequency shift with arene size is observed for the diarylpropanes VIa–VId. Evidently the flexible trimethylene linker permits optimal overlap for all of these excimers, whereas the more rigid urea linker restricts the extent of orbital overlap.
The excimers formed by the tertiary ureas IIb–IVb have low fluorescence quantum yields and short decay times at room temperature (Table 2). The observation that the excimer fluorescence intensity for IIb–IVb decreases upon warming MTHF solutions from above the glass transition temperature to room temperature suggests that an activated nonradiative decay process competes with excimer fluorescence. Flexibly linked dinaphthyl- and dianthrylalkanes are known to undergo intramolecular photocyclization.2,4,5 Thus, cyclization may account for the weak room temperature excimer fluorescence of IIb and IIIb. The photostability of the diarylureas remains to be investigated.
Phosphorescence
The diphenylureas Ia and Ic and the dinaphthylureas IIa and IIb display moderately intense structured phosphorescence at 77 K (Figs. 2, 3). The band shapes and triplet lifetimes are similar to those of the parent arenes and the 0,0 bands are shifted to slightly longer wavelength (Table 3). The weak phosphorescence of Ib is broader than that of either Ia or Ic, however, the band onset and maximum are at similar wavelengths. The observation of monomer rather than excimer phosphorescence for the tertiary as well as secondary diarylureas is consistent with observations for naphthalenophanes which display excimer fluorescence but monomer phosphorescence in low temperature glasses.13 No phosphorescence is observed for either the anthrylureas IIIa and IIIb or the pyrenylureas IVa and IVb. The parent arenes have very low intersystem crossing quantum yields as a consequence of large singlet–triplet energy gaps.21 Since the singlet behavior of these ureas is similar to that of the parent arene, it is not surprising that no phosphorescence is detected.Concluding remarks
N-Methylation of diarylureas results in a marked change in molecular conformation from extended for secondary diarylureas to folded for tertiary diarylureas. Whereas this pronounced difference in conformation has been exploited in molecular assembly and molecular recognition, its effects on molecular electronic structure and luminescence have not previously been investigated. The simplest of the diarylureas, diphenylurea, has been the most widely studied, but is essentially non-fluorescent in solution and hence the least interesting from our perspective. This is a consequence of the presence of a lowest n,π* singlet state which undergoes rapid intersystem crossing. The secondary diarylureas of the higher arenes all have lowest π,π* states and are strongly fluorescent at both 77 and 298 K. Their strong fluorescence and ability to serve as hydrogen bond donors and acceptors make them potentially attractive components of fluorescent sensors.The tertiary diarylureas of the higher arenes display monomer-like fluorescence in a rigid glass at 77 K but only excimer fluorescence in fluid solution. The absence of excimer fluorescence at 77 K is indicative of the occurrence of a large amplitude change in geometry upon excimer formation, presumably contraction of the arene–arene distance. The absence of monomer fluorescence in fluid solution is a consequence of the ground state conformational preference for a folded geometry. The ease of construction of tertiary diarylureas and homologous di- and triureas makes them attractive scaffolds for the investigation of electronic interactions between π-stacked chromophores.
Experimental
General methods
1H NMR spectra were measured on a Mercury 400 spectrometer. UV–VIS spectra were measured on a Hewlett–Packard 8452A diode array spectrometer using a 1 cm path length quartz cell. Total emission spectra were measured on a SPEX Fluoromax spectrometer. Low temperature spectra were measured either in a Suprasil quartz EPR tube (id = 3.3 mm) using a quartz liquid nitrogen cold finger dewar at 77 K or in an Oxford Cryogenics DN1704 cryostat fitted with an Oxford Instruments ITC4 temperature controller. Total emission quantum yields were measured by comparing the integrated area under the total emission curve at an equal absorbance and the same excitation wavelength as an external standard, 9,10-diphenylanthracene (Φf ≈ 1.0 at 77 K in MTHF).30 All emission spectra are uncorrected, and the estimated error for the quantum yields is ±10%. Fluorescence decays were measured on a Photon Technologies International (PTI) Timemaster stroboscopic detection instrument with a gated hydrogen or nitrogen lamp using a scatter solution to profile the instrument response function. Phosphorescence decays were measured on a PTI Timemaster phosphorescence timebased-detection instrument excited with a xenon-flash lamp as the excitation source. Nonlinear, least squares fitting of the decay curves employed the Levenburg–Marquardt algorithm as described by James et al.31 and implemented by the Photon Technologies International Timemaster (version 1.2) software. Goodness of fit was determined by judging the χ2 (<1.3 in all cases), the residuals, and the Durbin–Watson parameter (>1.6 in all cases). Solutions were degassed under vacuum (<0.1 Torr) through five freeze–pump–thaw cycles. ZINDO(S)/CI calculations were performed using Cache Release 4.4. Ground state structures were optimized with AM1 method implemented in MOPAC (version 94.10). Orbital diagrams were calculated using Hyperchem 5.0 or a Visualizer program implemented in the MOPAC program package. The diagrams obtained by the two methods are essentially the same.Materials
Triphosgene, triethylamine, aniline, 2-aminonaphthalene, 2-aminoanthracene, 1-aminopyrene and iodomethane are commercially available and were used as received. Anhydrous MTHF containing 200 ppm 2,6-di-tert-butyl-4-methylphenol (BHT, Aldrich) was distilled over sodium under a nitrogen atmosphere. N,N′-Diphenylurea (Ia) was recrystallized from DMF and water (10 ∶ 1) twice. N,N′-Dimethyl-N,N′-diphenylurea (Ib, Aldrich) was recrystallized from acetone and hexane (1 ∶ 1) twice.Preparation of the N,N′-diarylureas
To a solution of amine (10 mmol) in dichloromethane (20 mL), was added 2.2 equiv. of triethylamine, followed by 1.6 mmol. of triphosgene. After the mixture had been stirred for 10 min, the solvent was removed using a rotary evaporator. The solid residue was washed with water and recrystallized from DMF–water (10 ∶ 1). The resulting crystalline amide was washed with water and dried under vacuum.Preparation of tertiary N,N′-diarylureas
Into a solution of a N,N′-diarylurea (10 mmol) in 20 mL DMF was added dropwise 1.5 equiv. NaH in 10 mL DMF, followed by 1.5 equiv. of MeI. The mixture was stirred at room temperature until the starting material had been consumed (ca. 2–3 h), and was then mixed with water, and extracted with CH2Cl2. The organic layer was washed with water and dried with anhydrous potassium carbonate. After the solvent had been removed, the residue was purified by column chromatography using mixed solvent (acetone and hexane).Acknowledgement
Funding for this project was provided by NSF grants CHE-9734941 and CHE-0100596.References
- W. Klöpffer, in Organic Molecular Photophysics, ed. J. B. Birks, Wiley, London, 1973, p. 357 Search PubMed.
- W. Rettig, B. Paeplow, H. Herbst, K. Müllen, J.-P. Devergne and H. Bouas-Laurent, Intramolecular excimer formation in short- and long-chainlength di(9-anthryl) bichromophoric compounds and relation to ground state properties, New J. Chem., 1999, 23, 453–460 RSC.
- F. Hirayama, Intramolecular excimer formation. I. Diphenyl and triphenyl alkanes, J. Chem. Phys., 1965, 42, 3163–3171 CrossRef CAS.
- E. A. Chandross and C. J. Dempster, Intramolecular excimer formation and fluorescence quenching in dinaphthylalkanes, J. Am. Chem. Soc., 1970, 92, 3586–3593 CrossRef CAS.
- T. Haishi, N. Mataga, Y. Sakata, S. Misumi, M. Morita and J. Tanaka, Excimer fluorescence and photodimerization of anthracenophanes and 1,2-dianthrylethanes, J. Am. Chem. Soc., 1976, 98, 5910–5913 CrossRef CAS.
- F. C. De Schryver, N. Boens, H. Huybrechts, J. Daemen and M. DeBrackeleire, Photochemistry of bichromophoric compounds: scope and expectations, Pure Appl. Chem., 1977, 49, 237–247 CAS.
- K. A. Zachariasse and W. Kühnle, Intramolecular excimers with α,ω-diarylalkanes, Z. Phys. Chem., Neue Folge, 1976, 101, 267–276 Search PubMed.
- A. Tsuchida, T. Ikawa, T. Tomie and M. Yamamoto, Intramolecular pyrene excimer formation of 1,3-dipyrenylpropanes with full and partial ring overlaps, J. Phys. Chem., 1995, 99, 8196–8199 CrossRef CAS.
- P. Reynders, H. Dreeskamp, W. Kühnle and K. A. Zachariasse, Conformer analysis and intramolecular excimer formation with the meso and racemic diastereoisomers of 2,4-di(2-pyrenyl)pentane, J. Phys. Chem., 1987, 91, 3982–3992 CrossRef CAS.
- P. Reynders, W. Kühnle and K. A. Zachariasse, Ground-state dimers in excimer-forming bichromophoric molecules. 1. Bis(pyrenecarboxy)alkanes, J. Am. Chem. Soc., 1990, 112, 3929–3939 CrossRef CAS.
- S. Zeena and K. G. Thomas, Conformational switching and exciton interactions in hemicyanine-based bichromophores, J. Am. Chem. Soc., 2001, 123, 7859–7865 CrossRef CAS.
- P. Wahl, C. Krieger, D. Schweitzer and H. A. Staab, 1,8-Dipyrenylnaphthalenes: synthesis, molecular structure, and spectroscopic properties, Chem. Ber., 1984, 117, 260–276 Search PubMed.
- M. Yanagidate, K. Takayama, M. Takeuchi, J. Nishimura and H. Shizuka, Molecular conformation effects on the relaxation processes in the excited state of naphthalenophanes, J. Phys. Chem., 1993, 97, 8881–8888 CrossRef CAS.
- Y. Nakamura, R. Tsuihiji, T. Mita, T. Minowa, S. Tobita, H. Shizuka and J. Nishimura, Synthesis, structure, and electronic properties of syn-[2.2]phenanthrenophanes, J. Am. Chem. Soc., 1996, 118, 1006–1012 CrossRef CAS.
- W. Dannecker, J. Kopf and H. Rust, Crystal structure of N,N′-diphenylurea, Cryst. Struct. Commun., 1979, 8, 429–432 Search PubMed.
- M. C. Etter and T. W. Panunto, 1,3-Bis(m-nitrophenyl)urea: An exceptionally good complexing agent for proton acceptors, J. Am. Chem. Soc., 1988, 110, 5896–5897 CrossRef CAS.
- P. Ganis, G. Avitabile, E. Benedetti, C. Pedone and M. Goodman, Crystal and molecular structure of N,N′-diethyl-N,N′-diphenylurea, Proc. Natl. Acad. Sci. U. S. A., 1970, 67, 426–433 CAS.
- K. Yamaguchi, G. Matsumura, H. Kagechika, I. Azumaya, Y. Ito, A. Itai and K. Shudo, Aromatic architecture. Use of the N-methylamide structure as a molecular splint, J. Am. Chem. Soc., 1991, 113, 5474–5475 CrossRef CAS.
- H. Kagechika, I. Azumaya, K. Yamaguchi and K. Shudo, Sterical properties of N,N-dimethylurea moiety enhance formation of triply hydrogen-bonded complexes, Chem. Pharm. Bull., 1996, 44, 460–462 Search PubMed.
- R. K. Castellano, C. Nuckolls and J. Rebek, Jr., Transfer of chiral information through molecular assembly, J. Am. Chem. Soc., 1999, 121, 11156–11163 CrossRef CAS.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley-Interscience, London, 1970 Search PubMed.
- I. Carmichael and G. L. Hug, in Handbook of Organic Photochemistry, ed. J. C. Scaiano, CRC Press, Boca Raton, FL, 1989, Vol. I Search PubMed.
- M. C. Zerner, G. H. Loew, R. F. Kirchner and U. T. Mueller-Westerhoff, An intermediate neglect of differential overlap technique for spectroscopy of transition-metal complexes. Ferrocene, J. Am. Chem. Soc., 1980, 102, 589–599 CrossRef CAS.
- F. D. Lewis, W. Liu and T. L. Kurth, unpublished results.
- F. Vögtle, Cyclophane Chemistry, Wiley, Chichester, England, 1993 Search PubMed.
- C. R. Cantor and P. R. Schimmel, Biophysical Chemistry, W. H. Freeman, New York, 1969, Vol. II Search PubMed.
- R. V. Person, B. R. Peterson and D. A. Lightner, Bilirubin conformational analysis and circular dichroism, J. Am. Chem. Soc., 1994, 116, 42–59 CrossRef CAS.
- F. D. Lewis and W. Liu, Luminescence of N-arylbenzamides in low-temperature glasses, J. Phys. Chem., 1999, 103, 9678–9686 CrossRef CAS.
- F. D. Lewis and W. Liu, Temperature and conformation dependent luminescence of benzanilides, J. Phys. Chem., in the press Search PubMed.
- R. Li and E. C. Lim, Quantitative study of luminescence in aromatic and heteroraromatic molecules, J. Chem. Phys., 1972, 57, 605–612 CrossRef CAS.
- D. R. James, A. Siemiarczuk and W. R. Ware, Stroboscopic optical boxcar technique for the determination of fluorescence lifetimes, Rev. Sci. Instrum., 1992, 63, 1710–1716 CrossRef CAS.
- J. A. Settepani, R. T. Brown and A. B. Borkovec, Preparation of 1,3-diaryl-2-imidazolidinones. , J. Heterocycl. Chem., 1973, 10, 639–642 Search PubMed.
- H. C. Beachell, Chu-Pham-Ngoc-Son and Nguyen-Huu-Tinh, Pyrolysis of N-[β-(N″-phenylcarbamyl)ethyl]-N,N′-diphenylurea. Synthesis and properties of the decomposition product, 2-(phenylimino)-3-phenyloxazolidine and its analogs, J. Org. Chem., 1972, 37, 422–425 CrossRef CAS.
- L. F. Fieser and H. J. Creech, Conjugation of amino acids with isocyanates of the anthracene and 1,2-benzanthracene series, J. Am. Chem. Soc., 1939, 61, 3502–3509 CrossRef CAS.
- S. Tsuruoka, K. Naiki, E. Saito and K. Ota, Dialkyl dinaphthylureas, Yuki Gosei Kagahu Kyukaishi, 1959, 17, 144–148 ( Chem. Abstr. , 1959 , 53 , 11321 ) Search PubMed.
| This journal is © The Royal Society of Chemistry and Owner Societies 2002 |