Salt effects on the reactions of radical ion pairs formed by electron transfer quenching of triplet 2-methyl-1,4-naphthoquinone by amines. Optical flash photolysis and step-scan FTIR investigations
Marek
Mac†
* and
Jakob
Wirz
Institut für Physikalische Chemie, Universität Basel, CH4056-Basel, Klingelbergstrasse 80, Switzerland
First published on 2nd January 2002
Abstract
Electron transfer quenching of triplet 2-methyl-1,4-naphthoquinone by amines in acetonitrile was investigated by means of nanosecond flash photolysis and step-scan infrared spectroscopy. With 2,5-di-tert-butylaniline as the electron donor, proton transfer following the primary electron transfer process forms neutral radicals, which were identified by comparing the resulting transient IR absorption spectra with those obtained in the presence of phenol as the quencher. Addition of lithium perchlorate promotes the formation of free radical ions. The salt effect on the reactions of the nascent radical ion pairs formed by electron transfer is discussed in terms of Eigen theory. The quinone radical anion strongly associates with lithium cations. The salt retards the diffusional recombination of the free radical ions, as predicted by the Debye–Smoluchowski equation. The observed, predictable salt effects provide a useful diagnostic tool for elucidation of the reaction mechanism.
Introduction
Quinones are powerful electron acceptors1 and function as electron carriers in biological systems.2 In the excited triplet state quinones are reduced by neutral organic electron donors.3 The radical anions of the quinones and the radical cations of the corresponding organic donors were observed by flash photolysis and identified by comparison of the transient absorption spectra with those obtained by pulse radiolysis.4 The influence of solvent polarity on the electron transfer reaction is well established. Less attention has been paid to the influence of salts on the transient species formed by electron transfer.The yield of free ions depends on the free energy of back electron transfer, on the polarity of the solvent, and on the distance between the radicals in the nascent ion pair. Dissociation of the ion pair is inefficient, when back electron transfer processes predominate. Addition of lithium perchlorate (LiClO4), a redox-inert salt, accelerates the rate of charge separation,5 thereby enhancing the yield of free radical ions. For example, fluorescence quenching of 9,10-dichloroanthracene (DCA) by aromatic amines in acetone generates the radical anion of DCA and the radical cation of the aromatic donor.6 The radical ion pair decays by back electron transfer yielding the ground state or the triplet state of DCA, or it may dissociate into free radical ions that are readily detected by nanosecond laser flash photolysis (LFP). A related phenomenon is the quenching of exciplex fluorescence by lithium perchlorate:7 quenching rate constants increase with the extent of charge transfer in the exciplexes formed between excited 9,10-dicyanoanthracene and aromatic donors such as naphthalene and durene.
The above results show that inert salts influence the processes occurring in so-called ‘solvent separated radical ion pairs’ and ‘contact radical ion pairs’. In general, the inert salt does not affect the transient absorption spectra of the free radical ions. However, in experiments dealing with triplet state quenching of ketones by organic electron donors, addition of lithium perchlorate was noted to induce a substantial hypsochromic shift of the transient absorption spectra and to increase the lifetime of the ketyl radical anions.8
In this paper we report nanosecond LFP and step-scan FTIR experiments on the triplet state quenching of 2-methyl-1,4-naphthoquinone (MeNQ) by amines {1,4-diazabicyclo[2.2.2]octane (DABCO), 2,5-di-tert-butylaniline (DTBA), N,N-dimethylaniline (DMA)} and by phenol. Quenching rate constants are close to the diffusional limit, because the free energy of the primary electron transfer is strongly negative. The transient absorption spectra obtained by LFP are very broad due to overlapping absorption of the transient intermediates. Step-scan FTIR experiments permit identification of the transients that are formed when several primary photochemical processes compete.
Experimental
MeNQ, DABCO, and DTBA (all from Fluka) were used without purification. Acetonitrile (spectrograde, Fluka) and deuterated acetonitrile (Glaser) were dried using molecular sieve. DMA was distilled prior to use. The LFP equipment has been described.9 A 308 or 351 nm excimer laser was used for excitation to exclude photoionization of the quenchers. LFP experiments were carried out in both the presence and absence of air. Degassing was achieved by the freeze–pump–thaw technique.Infrared measurements were carried out on a Bruker IFS 66v/s Fourier transform infrared spectrometer equipped with a globar IR source, a KBr beam splitter and a nitrogen-cooled MCT detector. The step-scan technique10–12 was used for measurements of the transient IR spectra of MeNQ in the presence of quenchers. The spectral IR region was restricted to 2257–1130 cm−1 by using a cut on/cut off filter combination (LOT Oriel). The third harmonic from a Nd:YAG laser (355 nm) was used for excitation. The excitation energy was reduced to 4 mJ per pulse to avoid time-dependent signals from sample heating.12 Air-saturated solutions with an absorbance of 0.3 at the excitation wavelength were flowed through the cell (CaF2 windows, 200 μm path length) to avoid re-irradiation of transient intermediates. Comparison of the absorption spectra before and after the experiments indicated that the photoreactions were reversible.
Results
Optical flash photolysis
Excitation of MeNQ in acetonitrile leads to efficient and fast population of its triplet state (kISC = 1.1 × 1011 s−1),13 which decays with a lifetime of 230 ns in aerated, and of 3.6 μs in degassed solutions. The triplet–triplet (T–T) absorption band of MeNQ has a maximum at about 380 nm (Fig. 1, curve A). Addition of 8 mM DABCO quenches 3MeNQ within the 25 ns duration of the laser pulse. Instead of the exponential decay of the T–T absorption, a slower, second-order decay is now observed, and the maximum of the transient absorption is shifted to 400 nm (curve B). Furthermore, a second absorption band at 480 nm is observed, which decays with the same half-life as that at 400 nm. The absorption band at 400 nm is attributed to the radical anion of MeNQ, MeNQ−, and that at 480 nm to the radical cation of DABCO, DABCO+, by comparison with literature data.4 The observation of a clean second-order decay with the same half-life τ½ for both species is consistent with a charge recombination reaction MeNQ− + DABCO+ → MeNQ + DABCO, since the initial concentrations of the free radical ions must be equal. The half-life is defined as the time required for the decay of the transient absorbance to half of its initial value A0. Integration of the rate equation for such a scheme leads to a half-life τ½ = εd/(kA0), which is independent of the observation wavelength. The coefficient ε is the sum of the absorption coefficients of the transient intermediates at the wavelength of observation, d is the path length, and k is the second-order rate constant for recombination of the radical ions. Addition of 0.014 M LiClO4 causes a small hypsochromic shift of the radical anion band at 400 nm and enlarges its amplitude (curve C).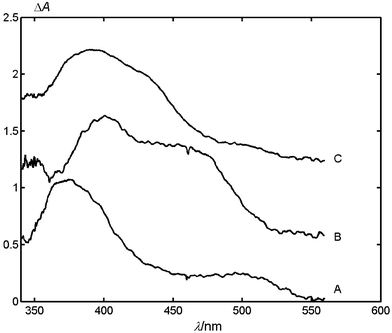 | ||
| Fig. 1 Normalized transient absorption spectra of (A) MeNQ, (B) MeNQ with 0.008 M DABCO, and (C) MeNQ with 0.008 M DABCO and 0.014 M LiClO4 in degassed acetonitrile taken 160 ns after excitation at 308 nm. The spectra B and C are shifted vertically for presentation purposes. | ||
The half-life for recombination increases upon addition of LiClO4 (Fig. 2). At high salt concentrations a plateau is reached.
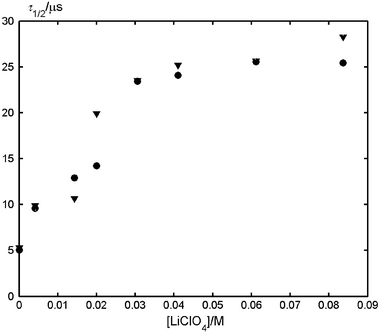 | ||
| Fig. 2 Salt concentration dependence of the half-lives of the radical anion of MeNQ (400 nm, ●) and of the radical cation of DABCO (470 nm, ▼) in air-saturated acetonitrile. The data were obtained by optical LFP. | ||
The rate constants are slightly higher in aerated than in degassed solutions, which may arise from quenching of the anion radical by oxygen. Addition of tetrabutylammonium perchlorate to a mixture of MeNQ and DABCO causes a small increase of the MeNQ radical anions' lifetime.
2,5-Di-tert-butylaniline (DTBA) also quenches the triplet state efficiently. The decay of the transient absorbance at 400 nm is slower than with DABCO as the quencher (τ½ = 3.54 μs with DABCO versus 23.5 μs with DTBA).
The transient absorption spectra of the mixtures containing DABCO or DTBA are very similar in the spectral region of 350–600 nm. The half-life of the species absorbing at 400 nm increases upon addition of LiClO4 to the mixture of MeNQ and DTBA. Similar results were obtained when N,N-dimethylaniline and phenol were used as triplet state quenchers of MeNQ. In general, optical flash photolysis gave broad, unstructured spectra. To identify the transient species formed in these systems, we turned to time-resolved (TR) FTIR.
Time-resolved FTIR measurements
Deuterated acetonitrile (CD3CN) was used as a solvent, because CH3CN absorbs strongly in the spectral region of 1250–2000 cm−1. Figs. 3A and B present the time-resolved IR difference spectra of MeNQ with DABCO (0.01 M) (A) without and (B) with 0.025 M LiClO4. Addition of the salt strongly lengthens the lifetime of the transient species formed by excitation of MeNQ, and results in a shift of some of the band maxima. The band at 1503 cm−1, whose decay obeys the second-order rate law with a half-life τ½ = 1.0 μs, is shifted to 1513 cm−1 (τ½ = 8.5 μs) upon addition of LiClO4, and the amplitude of the peak increases. Similar displacement is observed for the band absorbing at 1445 cm−1, which is shifted to 1458 cm−1. The position of the negative band at 1668 cm−1, which is due to depletion of MeNQ, is not affected by the addition of lithium perchlorate. Thus, the salt does not form a complex with MeNQ in the ground state.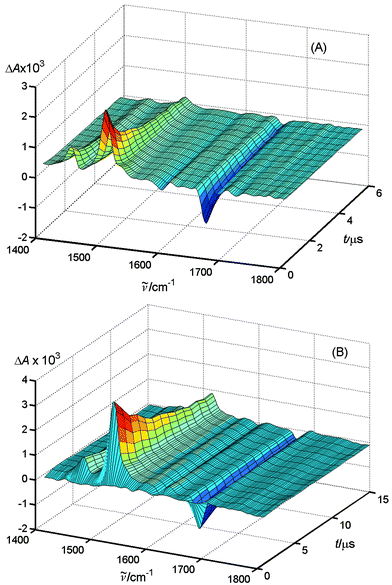 | ||
| Fig. 3 Time-resolved FTIR spectra of MeNQ + DABCO (0.01 M) (A) without and (B) with 0.25 M LiClO4 in CD3CN. | ||
In the presence of DTBA as a quencher, an additional absorption band is formed at 1536 cm−1 (Fig. 4), which exhibits a longer half-life, τ½ = 4.7 μs, than that at 1503 cm−1, τ½ = 1.3 μs. The half-life of the partial recovery of the band at 1668 cm−1 is equal to 4.1 μs, close to that of the decay at 1536 cm−1 (4.7 μs).
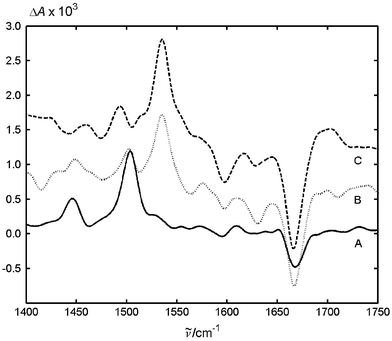 | ||
| Fig. 4 Difference IR absorption spectra of MeNQ with (A) 0.01 M DABCO, (B) 0.009 M DTBA and (C) 0.012 M phenol taken 500 ns after excitation. The spectra B and C are vertically shifted for clarity of presentation. | ||
Addition of lithium perchlorate to this system causes a shift of the band at 1503 to 1513 cm−1 and an increase of its amplitude (Fig. 5). The band at 1536 cm−1 is not affected. The half-lives at 1513 and 1536 cm−1 are also increased but the salt effect is more pronounced at 1503 cm−1: the half-life increases by a factor of 8.5 compared to 4.6 at 1536 cm−1. In the presence of phenol as a quencher, the most intense IR band is located at 1536 cm−1 (Fig. 5), and decays by second order kinetics (τ½ = 2 μs).
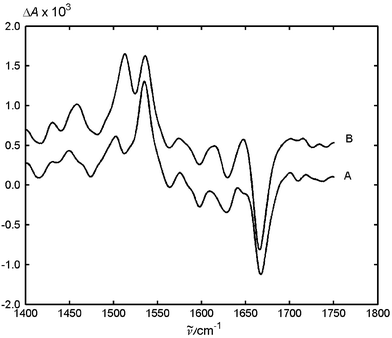 | ||
| Fig. 5 Difference FTIR absorption spectra of MeNQ with (A) 0.009 M DTBA, and (B) 0.009 M DTBA and 0.025 M LiClO4, taken 500 ns after excitation. Spectrum B is vertically shifted for presentation. | ||
Discussion
The reduction potential Eox of 2-methyl-1,4-naphthoquinone (MeNQ) is −0.71 V vs. SCE in acetonitrile, and the triplet state energy is 57.3 kcal mol−1 (2.485 eV).3 Hence, the triplet state of MeNQ will be quenched effectively by electron donors possessing oxidation potentials less than 1.77 V vs. SCE. The quenchers used here fulfil these conditions. The oxidation potentials vs. SCE of the amines are: DABCO (0.57 V),14 DTBA (0.94 V),15 DMA (0.81 V).16 The oxidation potential of phenol was estimated to be 1.5 V vs. NHE in water on the basis of the measured oxidation potential of phenolate and the acidity constants of phenol and phenoxyl radical.17 It is expected that the donating properties of phenol will be similar to those of anisole (1.76 V vs. SCE in acetonitrile16). Thus, phenol is the poorest electron donor among the quenchers used here.At the millimolar concentrations of the donors used, quenching of the triplet state of MeNQ is complete within the 25 ns duration of the laser pulse used for LFP. However, the resulting transient absorption spectra are diffuse and an assignment of the bands to the radical anion of MeNQ and the radical cation of the quenchers is difficult. Moreover, the spectra obtained with primary amine (DTBA) or phenol as quenchers resemble those obtained with DABCO and DMA, although formation of different transient species may be expected on the basis of the acidity of the radical cations formed and differences in the observed decay kinetics. With primary amines and phenol, proton transfer following the primary electron transfer step is likely. Evidence of such sequential processes in related systems is described briefly below.
Exciplex fluorescence of systems containing aromatic hydrocarbons (anthracene, pyrene, 9,10-dichloroanthracene) and aromatic amines in nonpolar solvents is observed only in the presence of tertiary amines, although secondary and primary amines also quench the fluorescence of such fluorophores.18 Proton transfer from the radical cation quenches the exciplex state (or contact radical pair) very effectively. Cation radicals of phenols are known to be strong acids with acidity constants pKa < 0;17a the acidity of the cation radical of aniline is significantly smaller with pKa = 7.17b By analogy, we expect that fast proton transfer from these donors follows electron transfer to MeNQ in the contact radical pair producing the ketyl radical MeNQH˙.
Scheme 1 shows the processes occurring after excitation of MeNQ at 351 (LFP) or 355 nm (step-scan FTIR) in the presence of the electron donors. Fast and efficient intersystem crossing populates the triplet state of MeNQ. Diffusional electron transfer generates radical ion pairs in the triplet state, which may dissociate into free radical ions. Back electron transfer in the primary radical ion pairs causing recovery of the ground state substrates is inhibited by spin restrictions. Thus, charge separation is efficient in the presence of quenchers that do not contain heavy atoms like bromine and iodine. However, the free radical ions may recombine and regenerate the ground state on subsequent diffusional encounter. With micromolar concentrations of the transients generated by flash photolysis, the transients' decays are in the microsecond region, suitable for observation by step-scan FTIR measurements. In the presence of primary amines or phenols, whose radical cations are strong acids, fast proton transfer scavenges the radical anion of MeNQ, and neutral radicals are formed. They recombine upon diffusional reencounter regenerating the starting materials, or may undergo further reactions.
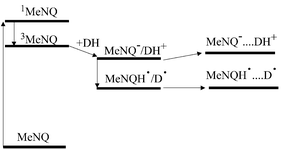 | ||
| Scheme 1 Processes following excitation of MeNQ in the presence of electron donors. | ||
Optical flash photolysis gave accurate information on the transient kinetics, but the diffuse transient spectra did not allow identification of the transient intermediates. More information was obtained by step-scan FTIR. In the presence of DABCO the difference spectrum in the region of 1400–1800 cm−1 shows strong bleaching at 1668 cm−1 and two much less intense, negative bands at 1627 and 1600 cm−1. These band positions and intensities correspond exactly to those in the IR spectrum of MeNQ, the first one to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, the second one to the quinone CC stretching vibration and the third one to CC stretching of the aromatic ring.19 After transfer of an electron from DABCO we expect changes in the electron density of MeNQ to be located mostly in the quinone moiety. Indeed, we observe a very intense absorption peak at 1503 cm−1 and a smaller one
at 1445 cm−1. The decays of these bands accurately follow a second-order rate law. The half-lives are the same for both decays, τ½
= 0.9 μs, and they correspond to that for recovery of the negative peak at 1668 cm−1, τ½
= 1.0 μs. Addition of 0.025 M lithium perchlorate does not change the position of the negative bands, but shifts the positions of the positive ones from 1503 and 1445 cm−1 to 1513 and 1458 cm−1. This indicates strong association of the radical anion with the lithium cation, which influences particularly the negatively charged CO bond and the quinone ring.
O stretching vibration, the second one to the quinone CC stretching vibration and the third one to CC stretching of the aromatic ring.19 After transfer of an electron from DABCO we expect changes in the electron density of MeNQ to be located mostly in the quinone moiety. Indeed, we observe a very intense absorption peak at 1503 cm−1 and a smaller one
at 1445 cm−1. The decays of these bands accurately follow a second-order rate law. The half-lives are the same for both decays, τ½
= 0.9 μs, and they correspond to that for recovery of the negative peak at 1668 cm−1, τ½
= 1.0 μs. Addition of 0.025 M lithium perchlorate does not change the position of the negative bands, but shifts the positions of the positive ones from 1503 and 1445 cm−1 to 1513 and 1458 cm−1. This indicates strong association of the radical anion with the lithium cation, which influences particularly the negatively charged CO bond and the quinone ring.
In the presence of DMA, depletion of the band at 1600 cm−1 is more pronounced, because the amine being oxidized also absorbs strongly at this position. The transient FTIR spectra (not shown) are very similar to those obtained in the presence of DABCO (Fig. 3). Again, the addition of lithium perchlorate changes the positions, half-lives, and amplitudes of the two positive peaks.
Addition of DTBA as the quencher modifies the transient absorption FTIR spectrum substantially (Fig. 4). In the region of 1700–1600 cm−1 we observe three intense negative peaks, which correspond to the ground state absorptions of MeNQ and of DTBA. The amplitudes of the negative bands at 1627 and 1600 cm−1 are stronger than those in the presence of DABCO and DMA. Besides the absorption band at 1503 cm−1, an intense one with a maximum at 1536 cm−1 is now observed. Addition of LiClO4 (Fig. 5) does not change the positions of the minima of the negative bands, which indicates that the quinone is not complexed by the salt. Also, the position of the positive band at 1536 cm−1 is not affected by the salt. The bands at 1503 and 1445 cm−1 are shifted to 1513 and 1458 cm−1, respectively. Moreover, the amplitude ratio of the absorption bands at 1536 and 1513 cm−1 is reduced by a factor of three. These observations indicate the presence of two different transient intermediates in this system. We assign the band at 1536 cm−1 to the ketyl radical MeNQH˙, and that at 1503 cm−1 to the radical anion MeNQ−. An explanation of the observed salt effects will be given below.
Recently, Burie et al.19 performed low temperature FTIR measurements for several quinones after irradiation in nonpolar media. They found no changes in the IR spectra upon irradiation at 10 K for ubiquinone, decyl ubiquinone, and 2,3-dimethyl-1,4-naphthoquinone. However, substitution by the unsaturated chains (isoprene units) in the 6-position of ubiquinone, or by phytyl chain (vitamin K) in the 3 position of 2-methyl-1,4-naphthoquinone causes significant changes in the IR difference spectrum upon irradiation at 10 K. The spectra of the substituted naphthoquinones possess an intense absorption band at 1535 cm−1, which is coincident with that observed by us in the presence of primary amine. Burie et al. attributed this band to a diradical, which is produced by intramolecular hydrogen transfer from the unsaturated chain to the proximal carbonyl bond of the quinone. The assignment was supported by EPR spectroscopy.
In another experiment, the triplet state of MeNQ was quenched by phenol. In this case the quenching could proceed by hydrogen atom transfer from phenol,3 or by sequential electron and proton transfer,17 resulting in the production of the ketyl radical. Indeed, the most intense peak is now located at 1536 cm−1 and is not affected by the addition of lithium perchlorate. With deuterated phenol, this band shifts from 1536 to 1528 cm−1. An additional, small peak located at 1513 cm−1 arises in the presence of the salt, which coincides with the absorption band of the complex between the lithium cation and the radical anion of MeNQ observed in the presence of amine quenchers. Formation of MeNQ− in the presence of inert salt suggests that the quenching of triplet ketones by phenol may proceed in a sequential fashion, viz. electron transfer followed by fast proton transfer within the radical pair,17 yielding ketyl radical and phenoxyl radical. This sequential process is well founded in light of the observed salt effect on systems with primary amines. Formation of MeNQ− is less effective with phenol as the quencher for reasons described in the next section.
Proton transfer and electron transfer
We observed two types of transient species, the radical anion of MeNQ, MeNQ−, and its protonated form, the ketyl radical MeNQH˙. Formation of the ketyl radical is dominant when phenol is used as the quencher. Sequential electron and proton transfer has been observed in various photogenerated radical ion pairs.20,21 Proton transfer occurs within the lifetime of the radical ion pair formed by electron transfer quenching, and this process escapes detection at 25 ns time resolution. In the systems containing primary amines, both MeNQ− and MeNQH˙ are observed in the TR FTIR measurements. The rate of proton transfer will depend on the acidity of the radical cations and on the distance between the two reactants after electron transfer. The acidity of phenol radical cation is expected to exceed that of aniline cations (phenol is a much stronger acid than anilines). Furthermore, due to the larger exothermicity of the electron transfer reaction with anilines (−0.835 vs. −0.01 eV for DTBA and phenol, respectively) we may expect that a smaller encounter distance is required for electron transfer quenching by phenol. Thus, the probability that proton transfer occurs in the radical pair will be larger for phenol than for aniline. Such a relationship between the encounter radius and the free enthalpy of electron transfer reactions was proposed and explored by Tachiya and Murata22 and by Mataga et al.23Bauscher and Mäntele24 reported FTIR studies of the reduction of several quinones by electrochemical methods. A thin-layer electrochemical cell was used to generate the reduced quinones, among them MeNQ in acetonitrile. The cyclic voltammograms showed two reduction waves for all quinones indicating electrochemical formation of semiquinone anions, Q−, and dianions, Q2−. Doubly reduced MeNQ was found to exhibit a band at 1380 cm−1 corresponding to stretching of the C–C aromatic bonds. We found a small peak at 1380 cm−1 in the presence of DMA and 0.25 M LiClO4, which is formed as the absorption band at 1513 cm−1, ascribed to Q−, decays (Fig. 6).
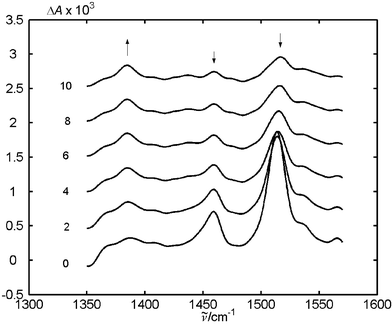 | ||
| Fig. 6 Transient FTIR spectra of MeNQ + DMA with LiClO4 in CD3CN. The spectra taken with delays of 2, 4, 6, 8, 100 μs after flashing were shifted for clarity. Arrows mark the significant temporal changes in the spectra. | ||
Salt effects
Addition of lithium perchlorate to the mixtures of MeNQ and the quenchers changes the properties of the transients. In the presence of DABCO and DMA we observe changes in the positions of the IR absorption bands that correspond to the radical anion of MeNQ, which indicate the formation of a complex between the lithium cation and MeNQ−. A small amount of LiClO4 (0.025 M) suffices to cause a hypsochromic shift of the absorption bands of MeNQ− indicating strong association. Optical LFP shows that the half-life of MeNQ− increases with increasing salt concentration. This dependence reaches a plateau at higher salt concentrations as depicted in Fig. 2.In the system containing the primary amine DTBA, addition of LiClO4 caused a change in relative band intensities (Fig. 5): the ratio of the band intensities due to MeNQH˙ (1536 cm−1) and MeNQ− (1503 cm−1) decreased threefold (the band of the complex [MeNQ−⋯Li+] is shifted to 1513 cm−1). The effect may be explained as follows: with primary amines, proton transfer follows the electron transfer step, and yields the contact (or solvent separated) radical ion pair. The pair separates into the free radical ions observed by LFP. They recombine on the microsecond timescale by second order kinetics. The separation rate of charged species depends strongly on ionic strength. For species of opposite charge increasing ionic strength accelerates charge separation as described by the Eigen equation [eqn. (1)]25 where w(r,μ) and β are given by eqns. (2) and (3).
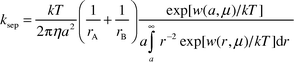
| (1) |
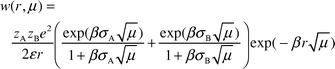
| (2) |
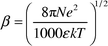
| (3) |
The second order rate constant for the recombination of the radical ions is expressed by the Debye–Smoluchowski equation [eqn. (4)].25
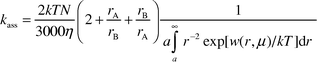
| (4) |
Here, k is the Boltzmann constant, η is the viscosity coefficient, N is Avogadro's constant, rA and rB are the radii, and a = rA + rB is the distance of closest approach of the reactants. The terms w(r,μ) and β are given by eqns. (2) and (3), in which zA and zB are the charges of ions A and B, ε is the relative permittivity of the solvent, e is the electron charge, μ is the ionic strength of the solution. Finally, σA and σB are the radii of the reactants A and B plus that of the dominant counterion. The solvated radii should be used in the calculation of ksep and kass instead of those of the bare ions.25
Fig. 7 shows the predictions of ksep and kass calculated for acetonitrile solutions with the appropriate parameters characterizing the system MeNQ + DABCO.
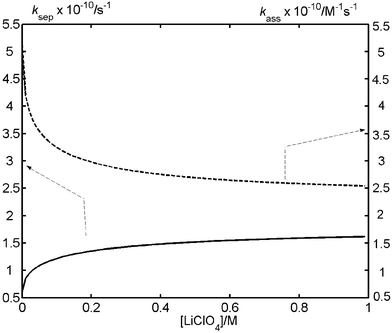 | ||
| Fig. 7 Dissociation (ksep) and association (kass) rate constants of the radical ion pair formed by triplet state quenching of MeNQ by organic quenchers in acetonitrile calculated as a function of salt concentration. Note that the dissociation rate constant is of first order while the association rate constant is of second order. The calculations were performed using eqns. (1)–(4) with the following parameters: rA (radius of MeNQ radical anion): 0.5 nm; rB (radius of organic radical cation): 0.3 nm; a (contact separation): 0.8 nm; ε (static relative permittivity of acetonitrile): 37.5; η (viscosity of acetonitrile): 0.0003 Pa s. The radii of the counterions, lithium cation and perchlorate anion, were taken as 0.1 and 0.226 nm, respectively. | ||
The theory predicts decreasing association rate constants and increasing dissociation rate constants as the salt concentration increases. The observed half-lives of the radical ions increase with increasing salt concentration (Fig. 2), which is in agreement with the predictions given by the Debye–Smoluchowski equation [eqn. (4)]. We also observe that the ratio of the IR band amplitudes assigned to the ketyl radical of MeNQ and that of the anion radical of MeNQ varies with salt concentration (decreases with increasing salt concentration). This is indirect evidence that the Eigen equation is fulfilled, because the increase of the dissociation rate constant with the addition of the inert salt decreases the probability for proton transfer following electron transfer within the radical pair.
Conclusions
Using step-scan FTIR methods we investigated the transient products formed upon reductive triplet state quenching of 2-methylnaphthoquinone (MeNQ) by amines and by phenol in acetonitrile. The IR transient absorption spectra of the radical anion and ketyl radical of MeNQ are consistent with those observed using different (bona fide) techniques for their production. Proton transfer follows the primary electron transfer step and competes with separation of the ion pair when the quencher radical cations are Brønsted acids. Consequently, both the quinone radical anion and the ketyl radical may be formed. The methods used here have insufficient time resolution to allow direct observation of the sequential electron and proton transfer steps, but the observed salt effects support such a mechanism. Addition of lithium perchlorate also affects the spectra and the lifetimes of the transient intermediates. Optical LFP and step-scan FTIR spectra indicate complex formation between the radical anion of MeNQ and the lithium cation.Acknowledgements
This work was supported by the Swiss National Science Foundation, grant No. 2000–061746.00. The authors would like to thank Professor S. Siebert and Dr C. Roedig from University of Freiburg (Germany) for introduction to the time-resolved infrared technique.References
- (a) R. Scheerer and M. Grätzel, Laser photolysis studies of duroquinone triplet state electron transfer reaction, J. Am. Chem. Soc., 1977, 99, 865–871 CrossRef CAS; (b) L. Loeff, J. Rabani, A. Treinin and H. Linshitz, Charge transfer and reactivity of nπ* and ππ* organic triplets, including anthraquinonesulfonates, in interactions with inorganic anions. A comparative study based on chemical Marcus theory, J. Am. Chem. Soc., 1993, 115, 8933–8942 CrossRef.
- R. Brudler and K. Gerwert, Step-scan FTIR spectroscopy resolves the Q(A)(−)Q(B) → Q(A)Q(B)(−) transition in Rb-sphaeroides R26 reaction centres, Photosynth. Res., 1998, 55, 261–266 CrossRef CAS.
- (a) I. Amada, M. Yamaji, S. Tsunoda and H. Shizuka, Laser photolysis studies of electron transfer between triplet naphthoquinones and amines, J. Photochem. Photobiol., A, 1996, 95, 27–32 Search PubMed; (b) I. Amada, M. Yamai, M. Sase and H. Shizuka, Laser flash photolysis studies on hydrogen atom abstraction from phenol by triplet naphthoquinones in acetonitrile, J. Chem. Soc., Faraday Trans., 1995, 91, 2751–2759 RSC.
- T. Shida, in Electronic absorption spectra of radical ions, Elsevier, Amsterdam, 1988, p. 311 Search PubMed.
- C. Chiorboli, M. T. Indelli, M. A. R. Scandola and F. Scandola, Salt effects on nearly diffusion controlled electron-transfer reactions. Bimolecular rate constants and cage escape yields in oxidative quenching of tris(2,2′-bipyridine)ruthenium(II), J. Phys. Chem., 1988, 92, 156–163 CrossRef CAS.
- M. Mac, Influence of lithium perchlorate on electron transfer processes in the systems: 9,10-dichloroanthracene and aromatic amines in acetone, J. Lumin., 1999, 81, 33–43 CrossRef CAS.
- M. Mac, P. Kwiatkowski and A. M. Turek, Quenching of exciplex fluorescence by lithium perchlorate in acetonitrile, Chem. Phys. Lett., 1996, 250, 104–110 CrossRef CAS.
- J. Mattay and A. M. Vondenhof, Top. Curr. Chem., 1991, 159, 220–252 Search PubMed.
- E. Hasler, A. Hörman, G. Persy, H. Platsch and J. Wirz, Singlet and triplet biradical intermediates in the valence isomerization of 2,7-dihydro-2,2,7,7-tetramethylpyrene, J. Am. Chem. Soc., 1993, 115, 5400–5409 CrossRef CAS.
- W. Uhmann, A. Becker, C. Taran and F. Siebert, Time-resolved FT-IR absorption spectroscopy using a step-scan interferometer, Appl. Spectrosc., 1991, 45, 390–397 Search PubMed.
- P. R. Griffiths and J. A. de Haas, in Fourier Transform Infrared Spectroscopy, John Wiley & Sons, NewYork, 1986, p. 268 Search PubMed.
- (a) C. Rödig, in Zeitaufgelöste Step-Scan-FTIR-Spektroskopie, University Dissertation, Freiburg, Breisgau, 1999 Search PubMed; (b) C. Rödig and F. Siebert, Errors and artifacts in time-resolved step-scan FT-IR spectroscopy, Appl. Spectrosc., 1999, 53, 893–901 Search PubMed.
- Y. Chiang, A. J. Kresge, B. Hellrung, P. Schünemann and J. Wirz, Flash photolysis of 5-methyl-1,4-naphthoquinone in aqueous solution: Kinetics and mechanism of photoenolization and of enol trapping, Helv. Chim. Acta, 1997, 80, 1106–1121 CrossRef CAS.
- E. Vauthey, P. Suppan and E. Haselbach, Free-energy dependence of the ion yield of photo-induced electron-transfer reactions in solution, Helv. Chim. Acta, 1988, 71, 93–99 CrossRef CAS.
- M. Mac and J. Wirz, unpublished results.
- K. Kikuchi, M. Hoshi, T. Niwa, Y. Takahashi and T. Miyashi, Heavy-atom effects on the excited singlet state electron-transfer reaction, J. Phys. Chem., 1991, 95, 38–42 CrossRef CAS.
- (a) S. Canonica, B. Hellrung and J. Wirz, Oxidation of phenols by triplet aromatic ketones in aqueous solution, J. Phys. Chem. A, 2000, 104, 1226–1232 CrossRef CAS; (b) Ling-Qin, G. N. R. Tripathi and R. H. Schuler, Radiation chemical studies of the oxidation of aniline in aqueous solution, Z. Naturforsch., A: Phys., Phys. Chem., Kosmophys., 1985, 40, 1026–1039 Search PubMed.
- T. Okada, I. Karaki and N. Mataga, Picosecond laser photolysis studies of hydrogen atom transfer reactions via heteroexcimer state in pyrene–primary and pyrene–secondary aromatic amines systems; role of ‘hydrogen bonding’ in interaction between amino group of donor and π-electron of acceptor in the heteroexcimer, J. Am. Chem. Soc., 1982, 104, 7191–7195 CrossRef CAS.
- J.-R. Burie, A. Boussac, C. Boullais, G. Berger, T. Mattioli, C. Mioskowski, E. Nebedryk and J. Breton, FTIR spectroscopy of UV-generated quinone radicals; evidence for an intramolecular hydrogen atom transfer in ubiquinone, naphthoquinone, and plastoquinone, J. Phys. Chem., 1995, 99, 4059–4070 CrossRef CAS.
- F. D. Lewis, Proton-transfer reactions of photogenerated radical ion pairs, Acc. Chem. Res., 1986, 19, 401–405 CrossRef CAS.
- M. Mac, J. Najbar and J. Wirz, Fluorescence quenching of derivatives of anthracene by organic electron donors and acceptors in acetonitrile. Electron and proton transfer mechanism, Chem. Phys. Lett., 1995, 235, 187–194 CrossRef CAS.
- M. Tachiya and S. Murata, New explanation for the lack of the inverted region in charge separation reactions, J. Phys. Chem., 1992, 96, 8441–8444 CrossRef CAS.
- T. Kakitani, A. Yoshimori and N. Mataga, Effects of the donor-acceptor distance distribution on the energy gap laws of charge separation and charge recombination reactions in polar solutions, J. Phys. Chem., 1992, 96, 5385–5392 CrossRef CAS.
- M. Bauscher and W. Mäntele, Electrochemical and infrared-spectroscopic characterization of redox reactions of p-quinones, J. Phys. Chem., 1992, 96, 11101–11108 CrossRef CAS.
- C. D. Clark and M. Z. Hoffman, Effect of solution medium on the rate constants of excited-state electron-transfer quenching reactions of ruthenium(II)-diimine photosensitizers, Coord. Chem. Rev., 1997, 159, 359–373 CrossRef CAS.
Footnote |
| † Present address: Wydział Chemii UJ, 30–060 Krakow, Ingardena 3, Poland. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2002 |