NMR studies on the axial chirality of ortho-substituted push–pull phenyl butadienes†
Manfred
Michalik
*a,
Thomas
Freier
a,
Helmut
Reinke
b and
Klaus
Peseke
b
aInstitut für Organische Katalyseforschung an der Universität Rostock, Buchbinderstr. 5-6, D-18055 Rostock, Germany
bFachbereich Chemie der Universität Rostock, Buchbinderstr. 9, D-18051 Rostock, Germany
First published on 28th November 2001
Abstract
1H NMR measurements of a series of ortho-phenyl substituted 3-aryl-2-cyano-5,5-bis(alkylthio)- (3h), 3-aryl-2-cyano-5-alkylthio-5-dialkylamino- (4h, 5) and 3-aryl-2-cyano-5,5-bis(dialkylamino)penta-2,4-dienenitriles (6) with prochiral groups showed the rotation about the C-3–C-aryl bond to be hindered within the NMR timescale. The activation parameters of this atropisomerization process are discussed with respect to steric effects of substituents. The rotation barriers correlate with bond lengths and angles as determined by X-ray structure analyses.
Introduction
In our studies on intramolecular mobility of the push–pull butadienes 1 and 2 (Scheme 1) we observed slow rotations about the C-2–C-3, C-3–C-4, C-4–C-5, and, for 2, the C-5–N bonds.1,2 Due to the π-electron interaction between the donor (Do) and acceptor groups and the diene double bond system these processes come into the NMR timescale and, therefore, are observable by dynamic NMR spectroscopy.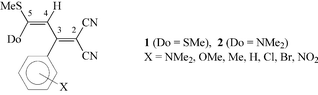 | ||
| Scheme 1 | ||
Furthermore, NMR spectra and X-ray crystal structure analyses1–3 of 1 and 2 have established the twisting of the phenyl ring out of the plane of the butadiene chain pointing out significant steric interactions with the donor and acceptor groups. Therefore, suitable phenyl substitution should enhance the steric barrier to rotation about the C-3–C-aryl bond giving rise to atropisomers with a chirality axis along this bond. In the case of perpendicular arrangement of the phenyl ring and the butadiene chain an unsymmetrical substitution in the ortho- or meta-position is necessary, whereas for the torsion angle θ ≠ 90°, chirality would also appear at para- or unsubstituted compounds (Fig. 1).
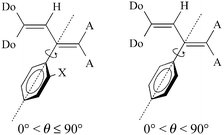 | ||
| Fig. 1 Rotation about the C-3–C-aryl bond. | ||
Referring to these considerations, the ortho-substituted push–pull butadienes 3–6 (Scheme 2) were prepared and studied by dynamic 1H NMR spectroscopy with respect to the stability of the chirality axis. With sufficiently high C-3–C-aryl rotation barriers, these compounds could be suitable precursors in asymmetric syntheses of heterocycles.4
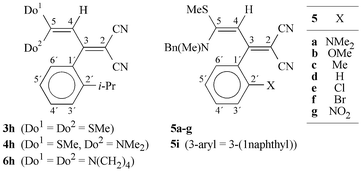 | ||
| Scheme 2 | ||
The synthesis of axially chiral compounds is directly connected with the question of stability of the chirality axes. For preparative separation of isomers at room temperature, an energy barrier ΔG≠ to rotation about the chirality axis of about 100 kJ mol−1 is necessary.5 Since the activation parameters describe the energy difference between ground states (GS) and transition states (TS), to enhance the barrier to C-3–C-aryl rotation, for instance by ortho-substitution, the influence of substituents on the GS as well as the TS has to be considered. A sterically bulky substituent does not inevitably lead to an increased energy barrier.6 In the case of strong electron-donating or -withdrawing groups electronic effects are of importance.7
Concerning the rotation about the C-3–C-aryl bond in 3–6, it was confirmed that the molecule is twisted in the GS similar to biphenyl derivatives.8 The phenyl ring is twisted out of the butadiene plane and, therefore, steric interactions with the substituent X in the GS should be of minor importance. The TS is supposed to be a nearly planar arrangement of the phenyl ring with the plane of the atoms C-2–C-3–C-4, whereas the remaining butadiene structure is twisted. In accordance with the rotation barriers found for the butadiene chain,2b the twisting of the donor part about the C-3–C-4 single bond should proceed more easily than the twisting of the nitrile groups about the C-2–C-3 double bond. Consequently, the rotation about the C-3–C-aryl bond would be characterized by two transition states, but, in the energy-lower state of the butadienes 3–6, the ortho-substituent will be moved about the more flexible donor side. These considerations are in agreement with results of quantum chemical calculations (see below). By dynamic NMR measurements this rotation process can only be observed if the enantiotopy and diastereotopy, respectively, of substituents of prochiral groups are changed (Scheme 3).9
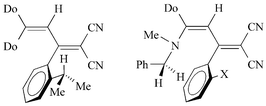 | ||
| Scheme 3 | ||
With that, exclusively the energy-lower step of the C-3–C-aryl rotation is detectable. Due to the planar transition state steric interactions cause higher TS energies and, therefore, increasing size of X should lead to higher rotation barriers.
Experimental
The 1H and 13C NMR measurements of 0.05 M solutions of 3–6 were performed on a Bruker ARX-300 spectrometer at 300.1 MHz and 75.5 MHz, respectively. The halogen containing solvents given in Table 1 were dried over molecular sieves or sodium sulfate and purified with basic aluminium oxide to remove acid impurities. For low-temperature measurements, argon was bubbled through the solution to remove impurities of paramagnetic oxygen. The probe temperature was measured by means of thermometer liquids.10 The exchange rates and activation parameters were obtained by CLSA (Complete Line Shape Analysis) of the prochiral groups i-Pr or Bn (Scheme 3) using the program DNMR5.11 The errors given in this paper refer to statistical errors of the linear regression ΔG≠vs.T in limits of 95% reliability (see ref. 12), for ΔG≠303K including a systematic temperature error of about ± 1 K. The real errors on ΔH≠ and ΔS≠ might be much larger. The true errors on ΔG≠303K approach the fitting errors, because the significant line-form alterations used for CLSA are in a relatively small range near room temperature and, therefore, the errors in ΔH≠ and ΔS≠ concerning the calculation of ΔG≠303K should to be neglected.| X | r V/Å | Solvent | ΔH≠/kJ mol−1 | ΔS≠/J mol−1 K−1 | ΔG≠303K/kJ mol−1 | |
|---|---|---|---|---|---|---|
| 3h | i-Pr | 1.96 | CDCl3 | 69.3 ± 0.6 | 2 ± 2 | 68.6 ± 0.2 |
| 4h | i-Pr | 1.96 | CDBr3 | 73.3 ± 0.5 | −11 ± 1 | 76.6 ± 0.3 |
| 6h | i-Pr | 1.96 | CDBr3 | 67.1 ± 1.0 | −30 ± 3 | 76.2 ± 0.4 |
| 5a | NMe2 | 1.63 | — | — | 61.8 ± 0.2 | |
| 5b | OMe | 1.52 | CD2Cl2 | 44.4 ± 1.1 | −31 ± 4 | 53.8 ± 0.4 |
| 5c | Me | 1.72 | CDCl3 | 58.3 ± 0.5 | −22 ± 2 | 64.9 ± 0.2 |
| 5e | Cl | 1.75 | CDCl3 | 60.9 ± 0.6 | −16 ± 2 | 65.8 ± 0.2 |
| 5f | Br | 1.85 | CDCl3 | — | — | 70.0 ± 0.3 |
| CDBr3 | 65.7 ± 0.4 | −12 ± 1 | 69.3 ± 0.2 | |||
| 5g | NO2 | 1.79 | CDCl3 | 63.6 ± 0.5 | −12 ± 2 | 67.2 ± 0.2 |
| 5i | — | — | CDBr3 | 69.3 ± 0.5 | −9 ± 1 | 72.1 ± 0.3 |
The X-ray diffraction results were obtained on a Bruker P4 four circle diffractometer with λ(Mo-Kα) = 0.71073 Å with graphite monochromator. After taking rotational photos and determining reasonable reduced cells a data collection was started in routine ω-scan. The structures were solved with direct methods (Bruker SHELXTL) and refined with the full-matrix least-squares method of SHELXL-97.13 All non-hydrogen atoms were refined anisotropically whereas the hydrogens were put into theoretical positions and refined according to the riding model.
The synthesis of butadienes 3h and 4h is described in ref. 1. The butadienes 5 were prepared according to ref. 1, 6h according to ref. 14. For all compounds, the experimental values of elemental analyses correspond to the calculated values within acceptable errors.
Synthesis of 3-aryl-5-benzyl(methyl)amino-2-cyano-5-methylthiopenta-2,4-dienenitriles (5) (general procedure)
2 mmol of the butadiene 3 in 8 mL of N-benzylmethylamine were stirred for 1–3 h at 100 °C. After cooling and addition of 10 mL of CHCl3 and water the reaction mixture was shaken with 2 mL (20 mmol) of conc. HCl, the organic phase was separated, washed three times with 10 mL of water and dried over Na2SO4. The CHCl3 was distilled off, the residue oil was dissolved in a small amount of EtOH and, after crystallization, recrystallized from EtOH.Synthesis of 2-cyano-3-(2-isopropylphenyl)-5,5-dipyrrolidinopenta-2,4-dienenitrile (6h)
1.5 mmol of 3h in 2 mL of pyrrolidine were refluxed for 2 h under stirring. After removing the solvent and dissolution in a little EtOH the formed crystals were recrystallized from EtOH. Yield: 55%, yellow–green needles, mp 203–204 °C. 1H NMR: δ (ppm) = 1.17 (d, 3H, CHCH3, J = 6.9 Hz), 1.34 (d, 3H, CHCH3, J = 6.8 Hz), 2.06 (br, 4H, CH2-3″), 3.26 (m, 1H, CHCH3), 3.62 (br, 4H, NCH2), 4.44 (s, 1H, H-4, s-cis-conformer (95%)), 5.26 (s, 1H, H-4, s-trans-conformer (5%)), 7.01 (m, 1H, H-6′), 7.12 (m, 1H, H-5′), 7.32 (m, 1H, H-4′), 7.35 (m, 1H, H-3′). 13C NMR: δ (ppm) = 23.5, 25.3 (CHCH3), 26.0 (CH2-3″), 29.0 (CHCH3), 45.8 (C-2), 50.9 (NCH2), 94.2 (C-4), 121.0 (CN), 125.4 (C-5′), 125.5 (C-3′), 128.1 (C-6′), 128.7 (C-4′), 139.1 (C-1′), 145.9 (C-2′), 160.4, 160.8 (C-3,5). MS (70 eV): m/z = 360 (M+).Results and discussion
The NMR detection of chirality was performed using prochiral isopropyl and benzyl groups. The methyl groups of the isopropyl moiety in 3h, 4h and 6h are nonequivalent at room temperature and show line-shape alterations on increasing the temperature. The CH2 signal of the benzyl group shows different line-shapes at room temperature depending on bulkiness of the ortho-phenyl substituents, e.g. unequivocal AB spectra for 5f, i and time-averaged signals for 5a, b. Examples for line-shape alterations of the isopropyl and benzyl signals are given in Fig. 2 and Fig. 3. In addition, rotations about the butadiene bonds could also be observed.1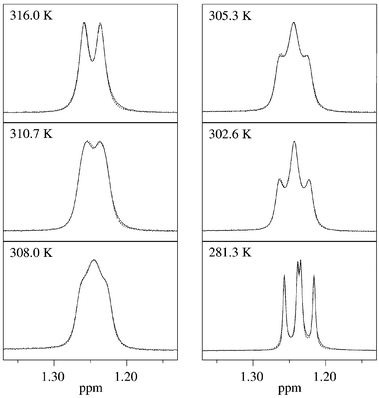 | ||
| Fig. 2 Temperature-dependent experimental and calculated (dotted lines) 1H NMR spectra of 3h (expanded plot of CH-Me signals). | ||
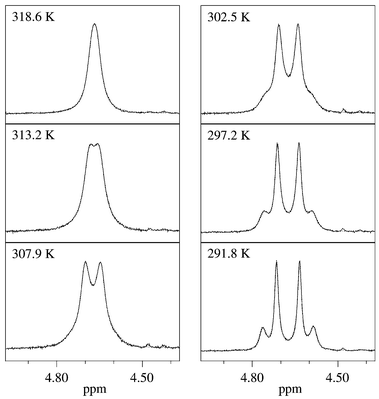 | ||
| Fig. 3 Temperature-dependent experimental and calculated (dotted lines) 1H NMR spectra of 5c (expanded plot of CH2 signal). | ||
C-3–C-aryl rotation barriers of butadienes 3h, 4h and 6h
The dependence of the C-3–C-aryl rotation barrier on substituents of the butadiene chain was studied for 3h, 4h and 6h (X = i-Pr in each case) (Table 1). As can be seen from ΔG≠303K, no compound possesses a stable chirality axis at room temperature. The significant increase 3h → 4h of about 8 kJ mol−1 on substitution of a methylthio by a dimethylamino group is due to the stronger donor effect of the amino group. It stabilizes the GS of the C-3–C-aryl bond in 4h by conjugative interactions, whereas it should not play a role in the TS owing to torsion of the donor part.The lack of a further increase in the rotation barrier on going from 4h to 6h can be explained by the different conformational structure of the butadiene chain. NMR chemical shifts and X-ray data prove the s-trans conformation for the butadienes 3h and 4h at room temperature, whereas for 6h the s-cis conformation was found to be favoured (see below).
In the GS the twisted phenyl ring in both conformations should exhibit comparable steric interactions with the donor as well as acceptor groups. In the planar TS, the phenyl group in the s-cis butadiene has a greater mobility because the steric interactions with the donor side are omitted. This effect could lead to a decrease of the rotation barrier and compensate the donor stabilizing of the ground state discussed above.
C-3–C-aryl rotation barriers of compounds 5
The influence of different ortho-phenyl substituents X on the C-3–C-aryl rotation has been studied for the monoaminobutadienes 5a–g including, in addition, the naphthyl-substituted compound 5i (Table 1).In the case of 5a the ΔH≠ and ΔS≠ values were not obtainable with sufficient accuracy because of additional line-broadening at low temperatures. For 5f in chloroform solution, the temperature range for the analysis was limited by the low boiling point of the solvent, therefore, ΔH≠ and ΔS≠ are not given. However, since line-shape alterations of the benzyl signals at room temperature could be observed and calculated for both compounds, the ΔG≠303K values are correct as given for the other butadienes 5 (Table 1).
For the unsubstituted compound 5d, activation parameters have not been determined due to further rotation processes and line-broadening in the temperature range concerned. Therefore, the assignment and determination of spectral parameters were impossible. However, the C-3–C-phenyl rotation is not necessarily observed (θ = 90°, Fig. 1).
The ΔG≠303K values of butadienes 5b–g are ranged corresponding to the size of substituents. A linear correlation between ΔG≠303K and van der Waals radii rV of the ortho-substituents X including 4h (Table 1, 5f in CDCl3) was found (Fig. 4, eqn. (1)).
| ΔG≠303K (kJ mol−1) = 50.66 rV − 23.03 (r = 0.997) | (1) |
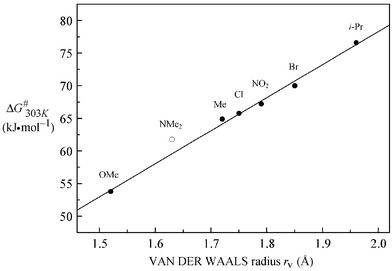 | ||
| Fig. 4 Correlation of ΔG≠303K of C-3–C-aryl rotation of the butadienes 5a–g (5f in CDCl3) and 4h with the van der Waals radii of the ortho-substituent X (○ not included (see text)). | ||
The atom radii of chlorine, bromine and oxygen15 (equal to methoxy group)8 and effective radii of the methyl, nitro and isopropyl group16 were included into the correlation. From the close correlation it can be concluded that the height of energy barriers of the C-3–C-aryl rotation is exclusively attributed to steric effects.
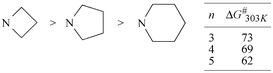
Compound 5a (X = NMe2) has not been included. The ΔG≠303K value of the C-3–C-aryl rotation is about 2 kJ mol−1 above the regression line. The deviation can be explained by the stronger donor capacity of the dimethylamino group. In this respect, further investigations17 on butadienes 5 with cyclic ortho-amino substituents X = N(CH2)n (n = 3–5) are worth mentioning. For these compounds, we also found unexpectedly high barriers to C-3–C-aryl rotations. The lower substituents show surprisingly the largest values resulting in the following range (ΔG≠303K in kJ mol−1, n = 3,4: 0.05 M in CDBr3, n = 5: 0.05 M in CD2Cl2) (Scheme 4).
 | ||
| Scheme 4 | ||
There are no van der Waals data for an 1-naphthyl group as in 5i. From the barrier to rotation an effective radius of 1.88 Å can be obtained which is in the range found for n-alkyl groups.16
By extrapolation to the radius of the H atom15 the ΔG≠303K value for the unsubstituted compound 5d can be estimated to be 38 kJ mol−1. In the same way, extrapolation for large substituents is possible. In the case of a tert-butyl group16 the ΔG≠303K can be estimated to be 100 kJ mol−1. The evaluation of dependence of the C-3–C-aryl rotation on substituent sizes leads to the conclusion that a chirality axis stable at room temperature can only be realized by substituents larger than the ortho-tert-butyl group or if a second ortho-substituent is introduced. However, the preparation of ortho,ortho′-substituted phenylbutadienes in the same way as the mono-ortho-substituted compounds 5 was not possible.
Crystal structures of butadienes 3h, 4h and 6h‡
In addition to the C-3–C-aryl rotation in solution the crystal structures of the isopropyl-substituted compounds 3h, 4h and 6h were studied (Tables 2 and 3). In the case of 3h two molecules exist in the asymmetric unit, which show distinct differences especially in the bond lengths. Otherwise the distances are in the ranges corresponding to the push–pull character.1| Atom distances/Å | Torsion angles/deg | |||||||
|---|---|---|---|---|---|---|---|---|
| C-2–C-3 | C-3–C-4 | C-4–C-5 | C-3–C-aryl | C-2–C-3a | C-3–C-4b | C-4–C-5c | C-3–C-aryld | |
| a θ(CN(Z)–C-2–C-3–C-4). b θ(C-2–C-3–C-4–C-5). c θ(C-3–C-4–C-5–Do). d θ(C-2–C-3–C-aryl–C-ortho-aryl-X). | ||||||||
| 3h | 1.366(4) | 1.427(3) | 1.364(4) | 1.500(3) | 5.8(4) | −175.1(3) | 0.1(5) | 89.8(4) |
| −177.6(2) | ||||||||
| 1.382(3) | 1.427(4) | 1.367(4) | 1.478(4) | 5.4(4) | −173.3(3) | −4.4(4) | 88.3(3) | |
| 177.1(2) | ||||||||
| 4h | 1.385(3) | 1.409(3) | 1.389(3) | 1.496(3) | 6.6(4) | −164.9(2) | 31.1(3) | −108.2(3) |
| −155.4(2) | ||||||||
| 6h | 1.431(4) | 1.354(4) | 1.459(4) | 1.503(4) | 6.3(5) | 5.7(5) | 64.9(4) | 84.8(3) |
| −117.4(3) | ||||||||
| 3h | 4h | 6h | |
|---|---|---|---|
| Sum formula | C17H18N2S2 | C18H21N3S | C23H28N4 |
| M w | 314.46 | 311.44 | 360.49 |
| Crystal size/mm | 0.66 × 0.40 × 0.12 | 0.78 × 0.70 × 0.40 | 0.48 × 0.20 × 0.15 |
| Colour | Yellow | Yellow | Yellowish |
| Crystal system | Triclinic | Monoclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
P |
| a, b, c/Å | 9.466(2), 11.727(2), 17.473(3) | 8.842(1), 11.368(1), 17.832(3) | 8.154(1), 12.041(1), 12.445(1) |
| α, β, γ/deg | 86.51(3), 76.31(3), 67.13(3) | 90.00, 96.24(1), 90.00 | 104.28(1), 102.50(1), 106.54(1) |
| V/Å3 | 1735.3(6) | 1781.8(4) | 1079.7(2) |
| Z | 4 | 4 | 2 |
| D c/g cm−3 | 1.204 | 1.161 | 1.109 |
| Abs. coefficient μ/mm−1 | 0.302 | 0.182 | 0.067 |
| F(000) | 664 | 664 | 388 |
| T Data collection/K | 298 | 298 | 298 |
| 2θ Range/deg | 3.78–45.00 | 4.26–45.00 | 4.22–44.00 |
| h, k, l Ranges | −5/10, −6/12, −18/18 | 0/9, 0/12, −19/19 | 0/8, −12/12, −13/12 |
| Reflections total | 3121 | 2342 | 2652 |
| Reflections obsd. (>2σI) | 2728 | 2013 | 1875 |
| R(int) | 0.0213 | 0.0305 | 0.032 |
| Parameters refined | 380 | 200 | 245 |
| Final R (all, obsd.) | 0.0411, 0.0360 | 0.0518, 0.0445 | 0.0829, 0.0552 |
| Final Rw (all, obsd.) | 0.0990, 0.0944 | 0.1257, 0.1189 | 0.1561, 0.1347 |
| GOF on F2 | 1.024 | 1.062 | 1.030 |
With enhanced donor–acceptor interaction a bonding balance along the butadiene chain is already observed for single donor-amino substitution. With double amino substitution the formal C-2–C-3 and C-4–C-5 double bonds become longer than the formal C-2–C-3 single bond. The most important structural differences to the unsubstituted butadienes3b are the Z-arrangement of the methylthio group in 4h and the s-cis conformation of 6h (Fig. 5). The s-cis conformation favoured for 6h was also found in solution (s-cis ∶ s-trans = 95 ∶ 5). Compared to 3h a strong increase of the torsion angle about the C-4–C-5 bond of the aminobutadiene 4h and 6h was observed, and for 4h additionally a C-3–C-aryl angle (Table 2).
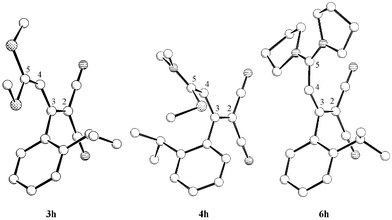 | ||
| Fig. 5 Molecular structures of the butadienes 3h, 4h and 6h (without H atoms). | ||
Molecular modelling
For the mechanism of the C-3–C-aryl rotation semiempirical AM1-calculations18 were made for compound 3h. First the GS geometry for the gas phase was optimized based on the crystal structure data (Fig. 5). The torsion angle of C-2–C-3–C-1′–C-2′ was about 90°. Starting with 90° we have varied θ(C-2–C-3–C-1′–C-2′) in 15° steps, in the range of TS in 5° steps, and optimized the geometries (Fig. 6).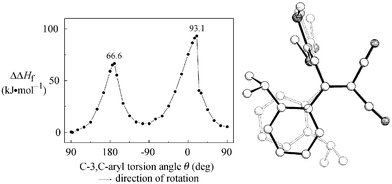 | ||
| Fig. 6 Enthalpy difference ΔΔHf = ΔHf − ΔHf,min (kJ mol−1) of 3h as a function of the torsion angle of C-2–C-3–C-1′–C-2′. AM1-optimized geometries (figure without H atoms) of both TS (plain figure, θ = −170°, ΔΔHf = 66.6 kJ mol−1; dotted figure, θ = 20°, ΔΔHf = 93.1 kJ mol−1). | ||
As already discussed two TS were calculated with planar arrangements of the phenyl ring towards the butadiene plane. For 3h, the energy for arrangement of X = i-Pr on the donor side is about 30 kJ mol−1 lower than for arranging on the acceptor side. The activation enthalpy ΔH≠ = ΔΔHf,max of the energetically lower TS (67 kJ mol−1) corresponds to the value obtained from dynamic NMR measurements (ΔH≠ = 69 kJ mol−1, Table 1). In Fig. 6 there are differences between the three GS minima. The reason should be that in the TS there is a nearly orthogonal arrangement of the butadiene chain, which is not retained in the following optimization cycles. However the enthalpies are only 5–9 kJ mol−1 above the minimum (s-trans conformation) and characterize the very low C-3–C-4 rotation barrier.
References
- T. Freier, M. Michalik, K. Peseke and H. Reinke, J. Chem. Soc., Perkin Trans. 2, 1999, 1265 RSC.
- (a) M. Michalik and K. Peseke, Sulfur Lett., 1986, 5, 55 Search PubMed; (b) M. Michalik, T. Freier, K. Zahn and K. Peseke, Magn. Reson. Chem., 1997, 35, 859 CrossRef CAS.
- (a) M. Michalik, Habilitationsschrift, University of Rostock, 1984 Search PubMed; (b) P. Dastidar, T. N. Guru Row and K. Venkatesan, Acta Crystallogr., Sect. B, 1993, 49, 900 CrossRef.
- (a) J. Liebscher, Sh. Jin, A. Otto and K. Woydowski, J. Heterocycl. Chem., 2000, 37, 509 Search PubMed; (b) K. Woydowski and J. Liebscher, Synthesis, 2000, 1444 CrossRef CAS; (c) A. Ali, V. U. Ahmad, J. Leistner and J. Liebscher, J. Chem. Soc., Perkin Trans. 1, 2000, 1897 RSC.
- L. Ernst, Chem. Unserer Zeit, 1983, 17, 21 Search PubMed.
- L. Lunazzi, A. Ticca, D. Macciantelli and G. Spunta, J. Chem. Soc., Perkin Trans. 2, 1976, 1121 RSC.
- (a) T. Drakenberg, R. Jost and J. Sommer, J. Chem. Soc., Chem. Commun., 1974, 1011 RSC; (b) T. Drakenberg, J. Sommer and R. Jost, J. Chem. Soc., Perkin Trans. 2, 1980, 363 RSC.
- G. Bott, L. D. Field and S. Sternhell, J. Am. Chem. Soc., 1980, 102, 5618 CrossRef CAS.
- (a) K. Mislow and M. Raban, Top. Stereochem., 1967, 1, 1 Search PubMed; (b) W. B. Jennings, Chem. Rev., 1975, 75, 307 CrossRef CAS; (c) G. Rembarz, E. Fischer and M. Michalik, Wiss. Z. Univ. Rostock, 1979, 28, 855 Search PubMed.
- (a) A. L. van Geet, Anal. Chem., 1968, 40, 2227 CrossRef CAS; (b) A. L. van Geet, Anal. Chem., 1970, 42, 679 CrossRef CAS.
- (a) D. S. Stephenson and G. Binsch, J. Magn. Reson., 1978, 32, 145 CAS; (b) D. S. Stephenson and G. Binsch, Quantum Chem. Program Exch., 1978, 10, 365 Search PubMed.
- R. E. Carter, T. Drakenberg and C. Roussel, J. Chem. Soc., Perkin Trans. 2, 1975, 1690 RSC.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, 1997 Search PubMed.
- K. Peseke, K. Zahn and M. Michalik, J. Prakt. Chem., 1994, 336, 357 CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- (a) M. Charton, J. Am. Chem. Soc., 1975, 97, 1552 CrossRef CAS; (b) M. Charton, J. Org. Chem., 1977, 42, 2528 CrossRef CAS.
- T. Freier, M. Michalik and K. Peseke, unpublished results.
- HyperChem 3, Hypercube, Inc., Gainesville, FL, USA, 1993 Search PubMed.
Footnotes |
| † Spectroscopic investigations on butadiene derivatives, Part 10. For Part 9, see ref. 1. |
| ‡ CCDC reference numbers 171037–171039. See http://www.rsc.org/suppdata/p2/b1/b107402b/ for crystallographic files in .cif or other format. |
| This journal is © The Royal Society of Chemistry 2002 |