Photoreduction induced by electron transfer from di- and trialkylamines to the triplet state of nitronaphthalenes in polar media
Received
(in Cambridge, UK)
13th August 2001
, Accepted 26th September 2001
First published on 28th November 2001
Introduction
The photoreduction of aromatic compounds by aliphatic amines is the subject of intensive investigations.1–8 The photoinduced reduction of the triplet state of 3,5,6-triphenyl-1,2,4-triazine and derivatives by triethylamine (TEA) and diethylamine (DEA) in several solvents has recently been studied by time-resolved spectroscopy.7 In oxygen-free solution these electron-deficient azaarenes are photochemically converted into dihydro derivatives which react back into the azaarenes on addition of oxygen. For phenazine in the presence of TEA the labile photoproduct is assigned to the corresponding 5,10-dihydrophenazine.8 A key step for the photoreduction of aromatic compounds in their triplet state is formation of the radical ion pair. For the photoinduced electron transfer from TEA to the triplet state of N-phenylnaphthalimides, two reduction steps, a primary and a secondary electron transfer take place.5 For azaarenes, however, the rate constant for triplet quenching (kq) is rather low and a possible two-step reduction is difficult to detect.
We became interested in searching for such a secondary, delayed step of radical formation in other systems with TEA. The photoreduction of nitro compounds, such as nitrobenzene9–20 and nitronaphthalenes,21–25 has been studied in the presence and absence of alcohols. Nitronaphthalenes are known to exhibit a high quantum yield of intersystem crossing (Φisc), whereas the fluorescence is negligible.21–31 For 1-methoxy-4-nitronaphthalene (MNN) the photosubstitution depends on the amine and the amount of water in acetonitrile.25 In the photoreduction of nitrobenzylidene malonic derivatives by TEA, nitroso compounds are postulated as labile intermediates prior to photoproducts from secondary excitation.32 The photoreduction of the triplet state of nitroaromatic compounds has been reviewed,29–31 but the specific role of aliphatic amines is still unclear. From the electroanalytical studies it is known that transfer steps of H-atoms lead from the radical anion of a nitro compound via a nitroso compound to the amino compound.33 It seemed desirable to get a deeper insight into the photoinduced reduction of fused nitroarenes by aliphatic amines.
1- and 2-Nitronaphthalene (1- and 2-NN) and MNN were chosen as study subjects for this work. Triplet quenching by TEA, DEA and 1,4-diazabicyclo[2.2.2]octane (DABCO) yields the radical anion of the nitronaphthalenes. The consequences of secondary reactions in the presence of TEA were studied by time-resolved spectroscopic and conductometric measurements in dry and in wet (5 M water) acetonitrile solutions.
Materials and methods
The nitronaphthalenes (Merck, Aldrich, K and K), DABCO, acetonitrile (Merck analytical grade for conductivity, otherwise Uvasol) and propan-2-ol were used as supplied. The purity of the nitronaphthalenes was checked by HPLC (>99%). TEA and DEA were purified by distillation. Water was from a Milli-Q system. The molar absorption coefficients of 1- and 2-NN at 354 nm are ε354
= 3 × 103 and 2 × 103 M−1 cm−1, respectively. The absorption spectra were monitored on a UV–vis diode array spectrophotometer (HP, 8453).7 A Nd laser (J. K. Lasers, pulse width 15 ns and energy <30 mJ) was used for excitation at 354 nm.34 The absorption signals were measured with two digitizers (Tektronix 7912AD and 390AD). Typically, absorbances of 1–2 were used for λexc, corresponding to concentrations of 0.5–1 mM. The fast (0.05–10 μs) and slow (5 μs–10 s) conductivity signals were measured by DC and AC bridges using the transient digitizers as reported elsewhere.34 For photoconversion the 366 nm line of a 200 W Xe–Hg lamp combined with an interference filter were used. The conversion was carried out after vigorous bubbling by argon prior to and during irradiation. For HPLC analyses a 125 × 4.6 mm Inertsil ODS-3 5 μm column was used, with MeOH–water 5 ∶ 1 as eluent. The quantum yield of decomposition (Φd) was determined using the Aberchrome 540 actinometer.35 All measurements refer to 24 ± 2 °C and deoxygenated solution, unless indicated otherwise.
Results
The maxima of the T–T absorption spectra, the triplet lifetime (τT) and the rate constant for quenching by oxygen of the three nitronaphthalenes (ArNO2) in acetonitrile are compiled in Table 1. For MNN τT is significantly shorter in the presence of water.25 The first-order decay rate constant (kobs), taken at 500 or 600 nm, shows a linear dependence on the TEA (open circles in Fig. 1), DEA and DABCO concentrations. The rate constant for triplet quenching, taken from the slope of these plots, is kq
= (1.4–1.8) × 1010 M−1 s−1 in acetonitrile (Table 2). In water–acetonitrile mixtures (1 ∶ 9, vol.) the kobs values depend also linearly on [TEA] (full circles in Fig. 1), but the kq values are generally much lower than in neat acetonitrile, ≊2 × 109 M−1 s−1, whereas the spectra are very similar. Markedly lower kq values on addition of water to acetonitrile have also been reported for azaarenes and the MNN–TEA system.7,25 The values for the latter are 1.4 × 1010 and 2.5 × 109 M−1 s−1 for water concentrations of 0 and 14 M, respectively. The reason may be hydrogen bonding between water and the amine lone pair, making the latter less oxidizable.
Table 1 T–T absorption maxima of nitronaphthalenes, triplet lifetimea and rate constant for quenching by oxygen
| Compound |
λ
TT
b/nm |
τ
T
c/μs |
109koxd/M−1 s−1 |
|
In argon-saturated acetonitrile.
Main band underlined.
At low pulse intensity.
Using air- and oxygen-saturated solution.
Taken from ref. 25.
|
| 1-NN |
400, 560 |
10 |
1.4 |
| 2-NN |
480 |
8 |
1.3 |
| MNN |
420, 630 |
10 |
1.2/2e |
Table 2 Rate constant for quenching by amines, absorption maximum and half-live of the resulting secondary transients and quantum yield of decompositiona
| Compound |
Donor |
109kq/M−1 s−1 |
λ
rad
b/nm |
t
1/2/ms |
Φ
d
c
|
|
In argon-saturated acetonitrile.
Using [amine] = 0.1–10 mM.
Obtained by absorption and/or HPLC using λirr
= 366 nm and [amine] = 10 mM.
In parentheses: plus water (5 M).
Air-saturated solution.
Taken from ref 25.
|
| 1-NN |
TEA |
16 (2)d |
395 (390)d |
0.3–2 (10)d |
0.08 [<0.002]e |
| |
DABCO |
18 (2) |
395 |
<0.05 |
| 2-NN |
TEA |
15 |
390 |
0.5–3 [<0.01]e |
0.08 |
| |
DEA |
15 |
390 |
0.4–2 |
0.05 |
| |
DABCO |
18 |
395 |
<0.04 |
| MNN |
TEA |
14 |
395 |
0.5–3 |
0.1 [<0.002] |
| |
DEA |
|
395f |
| |
DABCO |
18 |
395 |
<0.05 |
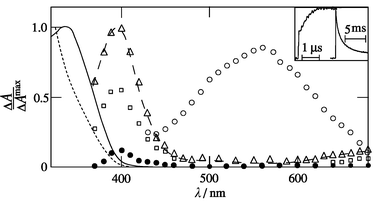
![Rate constant for triplet decay of 1-NN at 560 nm (circles) and of grow-up in absorption at 400 nm (triangles) vs. [TEA] in dry acetonitrile (open symbols, a, b) and in acetonitrile–water (9 ∶ 1, vol.) (filled symbols, c); insets: kinetics at 400 nm (left) and 560 nm (right) ([TEA], a: 2 mM, b,c: 20 mM).](/image/article/2002/P2/b107328c/b107328c-f1.gif) |
| | Fig. 1 Rate constant for triplet decay of 1-NN at 560 nm (circles) and of grow-up in absorption at 400 nm (triangles) vs. [TEA] in dry acetonitrile (open symbols, a, b) and in acetonitrile–water (9 ∶ 1, vol.) (filled symbols, c); insets: kinetics at 400 nm (left) and 560 nm (right) ([TEA], a: 2 mM, b,c: 20 mM). | |
In the presence of TEA the initial T–T absorption spectrum in argon-saturated acetonitrile is unchanged (see circles in Figs. 2, 3a and 4a), but the triplet lifetime is reduced. The triplet was detected (by ns time resolution) only when [TEA] was below 10 mM. This was also the case for DEA. A 50% triplet quenching by the three amines is achieved at rather low concentrations: [amine]1/2
= 5 μM, taking τT
= 10 μs and kq
= 2 × 1010 M−1 s−1. After disappearance of the 1-NN triplet, a remaining transient absorption with maximum (λrad) at 395 nm was detected in the presence of DABCO (Fig. 5). This species, which decays by second-order kinetics with a first half-life of t1/2
= 10–50 μs, is assigned to the radical anion (ArNO2˙−). Electron transfer to the triplet state (3*ArNO2) of the nitronaphthalenes and electron back transfer to the DABCO radical cation (DH2˙+, Scheme 1) occurs via reactions (1)–(4).
 |
| | Fig. 2 Transient absorption spectra of 1-NN in argon-saturated acetonitrile in the presence of TEA (5 mM) at 20 ns (○), 0.1 μs (□), 1 μs (△) and 10 ms (●) after excitation by 354 nm pulses; inset: kinetics for grow-in and decay at 400 nm; ground state prior to (full) and after conversion (broken) with λirr
= 366 nm. | |
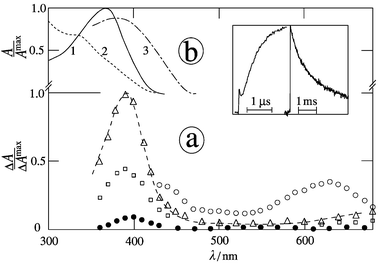 |
| | Fig. 3 (a) Transient absorption spectra of MNN in the presence of TEA (5 mM) in argon-saturated acetonitrile at 20 ns (○), 0.1 μs (□), 1 μs (△) and 10 ms (●) after the 354 nm pulse; inset: kinetics for grow-in and decay at 400 nm; (b) ground state spectra prior to (1) and after conversion (2) with λirr
= 366 nm and (3) after addition of oxygen. | |
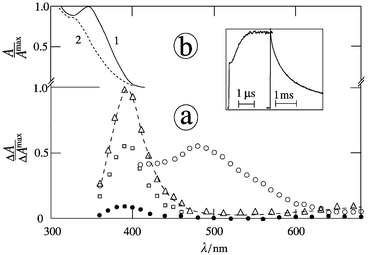 |
| | Fig. 4 Transient absorption spectra of 2-NN in the presence of TEA (5 mM) in argon-saturated acetonitrile at 20 ns (○), 0.1 μs (□), 1 μs (△) and 10 ms (●) after the 354 nm pulse; inset: kinetics for grow-in and decay at 400 nm; (b) ground state spectra prior to (1) and after conversion (2) with λirr
= 366 nm. | |
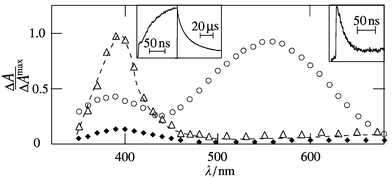 |
| | Fig. 5 Transient absorption spectra of 1-NN in argon-saturated acetonitrile in the presence of DABCO (1 mM) at 20 ns (○), 1 μs (△) and 50 μs (♦) after excitation by 354 nm pulses; insets: kinetics at 400 nm (left) and 560 nm (right). | |
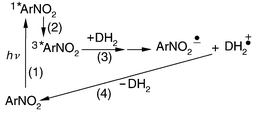 |
| | Scheme 1 | |
Secondary transients
Examples of the absorption spectra of the secondary transients of 1-NN, 2-NN and MNN in acetonitrile in the presence of TEA are presented in Figs. 2, 4a and 3a, respectively. The species subsequent to the nitronaphthalene triplet state should be assigned (at least the major part) to the nitronaphthalene radical anion on the basis of the comparison of the spectra obtained with DABCO (Table 2) and the second-order decay kinetics. For 1-NN in water the molar absorption coefficient of the radical anion is ε380
= 1.0 × 104 M−1 cm−1.27 At higher TEA concentrations (1–10 mM) the transient absorption at λrad contains two similar parts; one is formed directly by triplet quenching and the second one is formed delayed (Figs. 2, 3a and 4a). The absorption spectra of both parts are the same, having λrad
= 395 nm. The second delayed part, increasing within 1 μs by a factor of 1–3, was also observed for DEA at higher concentrations, in contrast to DABCO. For [H2O] = 5 M and an appropriate TEA concentration each absorption signal consists also of two subsequently formed parts which have essentially the same spectra (Table 2).
Continuous irradiation
Continuous UV irradiation at 366 nm of 1- or 2-NN and MNN in argon-saturated acetonitrile in the presence of TEA (1–10 mM) leads to decomposition of substrate and no absorption in the visible region (Figs. 2, 3b and 4b). The quantum yields of decomposition (Φd) in the presence of TEA are compiled in Table 2. A maximum Φd value in most cases was achieved under careful exclusion of oxygen and when [TEA] is larger than 0.1 mM. Primary photoproducts are indicated by isosbestic points at 272, 337 nm and 275, 335 nm for 1-NN and MNN, respectively. They are not present any more when photolysis of the photoproduct(s) becomes noticeable. The decomposition of the substrate was also measured by HPLC; for 1- and 2-NN the amount of products (observed at 350 nm) for 50% conversion is less than 10%. Moreover, addition of oxygen to a solution containing the photoproduct of MNN leads to a different product as indicated by the absorption spectrum with red-shifted maximum (curve 3 in Fig. 3b). The latter effect is also present for 1- or 2-NN but is smaller.
Nitronaphthalenes generally do not fluoresce. Formation of the photoproduct(s) of MNN in the presence of TEA (or DEA), however, goes along with a fluorescence with an emission maximum located at 470 nm and an excitation maximum is at 330 nm. The signal increases (>100 fold) and is proportional to the conversion (not shown). Corresponding effects were not observed with the 1- or 2-NN–TEA systems. Since the fluorescence cannot originate from MNN itself, it is coming from a product. Since 1-methoxynaphthalene is unlikely as one possibility 1-hydroxyamino-4-methoxynaphthalene may be considered.
Conductivity in the presence of amines
A conductivity increase (Δκ is positive for increasing conductivity) was observed in the pulsed experiments for nitronaphthalenes in argon-saturated neat acetonitrile in the presence of DABCO. The Δκ signal increases within the 1 μs range (inset a in Fig. 6) and decays by second-order kinetics with half-lives in the 0.02–0.1 ms range depending on the laser intensity (Table 3). Similar results for TEA at low concentrations (<0.1 mM) are in agreement with photoinduced electron transfer and back electron transfer. The time dependences of representative cases in the presence of TEA (>0.1 mM) are given for 1-, 2-NN and MNN (Fig. 6). The rate constant of the fast increase depends linearly on the TEA concentration and the signal approaches a maximum at TEA concentration larger than [TEA]1/2.
Table 3 Relative signal amplitude and half-life obtained from transient conductivity of nitronaphthalenesa
| Compound |
Donor |
Δκ/Δκmaxb |
t
1/2/ms |
|
In argon-saturated acetonitrile, using [amine] = 0.2–10 mM.
Taken under normalized conditions.
In parentheses: plus water (5 M).
Oxygen-saturated acetonitrile: t1/2 <0.001.
|
| 1-NN |
TEA |
1.0 (0.9)c |
0.4–2 (>30)c |
| 2-NN |
TEA |
0.9 |
0.5–2 (>20) |
| |
DEA |
0.8 |
0.4–2 (>10) |
| |
DABCO |
0.8 |
0.03–0.1(<0.1) |
| MNN |
TEA |
0.8 |
2–8 (>40)d |
![Conductivity signals as a function of time of 1-NN (circles), MNN (squares) and 2-NN (triangles) in the presence of TEA (0.01 M) in argon-saturated acetonitrile (open) and acetonitrile–water (9 ∶ 1, full); insets: increase for 1-NN in acetonitrile ([TEA] = 1 mM a: argon and a′: oxygen), acetonitrile–water (9 ∶ 1, b: [TEA] = 1 mM and b′: 0.1 mM) and decay in dry (c) and in wet (d) acetonitrile.](/image/article/2002/P2/b107328c/b107328c-f6.gif) |
| | Fig. 6 Conductivity signals as a function of time of 1-NN (circles), MNN (squares) and 2-NN (triangles) in the presence of TEA (0.01 M) in argon-saturated acetonitrile (open) and acetonitrile–water (9 ∶ 1, full); insets: increase for 1-NN in acetonitrile ([TEA] = 1 mM a: argon and a′: oxygen), acetonitrile–water (9 ∶ 1, b: [TEA] = 1 mM and b′: 0.1 mM) and decay in dry (c) and in wet (d) acetonitrile. | |
The half-lives (inset c in Fig. 6) increase by a factor of ca. 5–8 when the laser intensity is decreased to one tenth. This holds also in the presence of 5 M water, where the positive signal also reaches a plateau value in the 0.1–1 μs range (insets b, b′ in Fig. 6), but the half-lives are typically an order of magnitude longer (inset d in Fig. 6). Virtually no further increase of the conductivity signal was registered in neat acetonitrile even at higher TEA concentrations of 2–8 mM. However, indication for such an effect is found for 1-NN in acetonitrile–water (9 ∶ 1) (Fig. 6, inset b). The conductivity signal under normalized conditions (constant laser intensity and absorbance at 354 nm, [amine] = 10 mM) is not specific for the types of either nitronaphthalene or amine; 5 M water has virtually no effect (Table 3). The presence of oxygen has no marked effect on the initial conductivity signal under conditions of sufficient triplet quenching using [TEA] = 1–10 mM, but the decay in the 0.1–10 ms range is significantly decreased. Nevertheless, a small component remains in the 0.1–10 ms range in oxygen-saturated acetonitrile, this being significantly higher in the presence of 5 M water.
Discussion
Some properties of the triplet state of 1- or 2-NN have been reported.22–26 The triplet energy of 1- and 2-NN is ET
= 240 kJ mol−1.24,26,29 The Φisc values of 1- and 2-NN in solution at room temperature are 0.6–0.8 and similar in glassy media at −196 °C.29,30Φisc is also substantial for MNN in acetonitrile, where τT
= 4–5 μs and kox
= 2 × 109 M−1 s−1, but in contrast to the other nitro compounds the triplet lifetime becomes significantly smaller on addition of water.25
Decay of the radical anion of the three nitronaphthalenes in the case of DABCO occurs via back electron transfer (4). In principle, the radical anion could be converted into Ar˙NO2H, the conjugate acid.
| | |
ArNO2˙−
+ H2O ⇄ Ar˙NO2H + OH−
| (5) |
For the nitroaromatics in acetonitrile and mixtures of acetonitrile with water (pH >9), however, this protonation may be excluded. From pulse radiolysis studies of nitrobenzene in aqueous solution the pKa value of 3.2 is known for equilibrium (5).36 The observed transient with a characteristic maximum at 390 nm, second-order decay kinetics and a rather short half-life (Table 2) is assigned to the radical anion of the nitronaphthalene, formed by electron transfer from DABCO to the triplet state (Scheme 1).
Closer inspections of the delayed formation of the longer lived intermediates show that the absorption spectra are very similar or even identical and that the build-up kinetics depend on the TEA concentration.
| | |
Et3N˙+
+ Et3N ⇄ Et2N–˙CHMe + Et3NH+
| (6) |
The likely possibility which should dominate at higher [TEA] is forward reaction with a second TEA molecule to form the triethylammonium cation (Et3NH+
= DH3+ in Scheme 2) and the α-aminoethyl radical (D˙H) in equilibrium (6). For the photoinduced electron transfer from TEA step (6) was found to occur with k6
= 5.7 × 109 and 1.9 × 109 M−1 s−1 for 1,8- and 2,3-N-phenylnaphthalimide, respectively.5 This is consistent with the finding that the ratio of rate constants of forward to backward reactions is approximately 100,6i.e. the Et2N–˙CHMe radical will not react back under the applied conditions using [TEA] = 1–100 mM. For the case of DEA, Et2N–˙CHMe has to be replaced by the EtHN–˙CHMe radical. It is worth mentioning that with DEA the same k6 value as with TEA was found with the N-phenyl-1,8-naphthalimide system.5
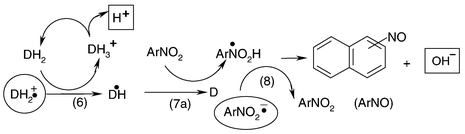 |
| | Scheme 2 | |
Formation of secondary radicals derived from nitronaphthalenes can be described by reactions (7a)–(7c), where Et2N–CH(Me)–O–N(O˙)–Ar denotes an adduct radical.
| |
Et2N–˙CHMe + ArNO2
→ Et2NCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2
+ Ar˙NO2H CH2
+ Ar˙NO2H
| (7a) |
| | |
Et2N–˙CHMe + ArNO2
→ Et2N+CHMe + ArNO2˙−
| (7b) |
| | |
Et2N–˙CHMe + ArNO2
→ Et2N–CH(Me)–O–N(O˙)–ArAr
| (7c) |
The half-life of the growth of absorption at λrad decreases with [TEA] from a typical value of 2 μs at 2 mM TEA in acetonitrile to 100 ns at 20 mM (Fig. 1). This gives a rate constant of krad
= 5 × 108 M−1 s−1. In aqueous acetonitrile (5 M water) the grow-up is slower and krad
= 1 × 108 M−1 s−1 can be estimated. For the reaction of α-aminoalkyl radicals with benzil in di-tert-butyl peroxide, rate constants of k7
= 1.8 × 109 and 3.3 × 109 M−1 s−1 have been reported for TEA and DEA, respectively.3 For the MNN–TEA system the k7 value is 3.9 × 109 M−1 s−1.25 The possibilities for radical–radical termination are reactions (8)–(11) (Scheme 2).
| | |
ArNO2˙−
+ Ar˙NO2H → ArNO2
+ ArNO + OH−
| (8) |
| | |
ArNO2˙−
+ Ar˙NO2H + H2O → (ArNO2H)2
+ OH−
| (8′) |
| | |
2 × Ar˙NO2H → ArNO2
+ ArNO2H2
| (9) |
| | |
2 × Ar˙NO2H → (ArNO2H)2
| (9′) |
| | |
2 × Et2N–˙CHMe → Et3N + Et2NCHCH2
| (10) |
| | |
Et2N–˙CHMe + X–Ar′–NO2˙−
→ HAr′–NO2
+ X−
+ Et2NCHCH2
| (11) |
Reactions (10) and (11) would lead to N,N-diethylvinylamine (D in Scheme 2) which has to be replaced by N-ethyliminoethane for the case of DEA. Reaction (11) describes elimination of a substitutent, e.g. X = OMe in the case of MNN.25Reaction (10), which competes with reactions (7) and (11), is not accessible by our spectroscopic analysis. Photodimers from reactions (8′) and (9′) could be detected for other systems,1 but for the cases of nitronaphthalenes no indication for dimers was found and disproportionation competes probably successfully. A self-termination reaction of two radical anions is expected to be very slow, based on pulse radiolysis studies of nitrobenzene in aqueous solution.35 Step (8) refers to formation of the nitroso compound (ArNO) and should be distinguished from the dehydration of ArNO2H2, which may be formed via (9). Radical disproportionation via a dihydro derivative is known for naphthalene,37 but is unlikely for nitro compounds and cannot account for the charge separation detected by conductivity (Fig. 6).
The photoinduced electron transfer from amines to the nitronaphthalenes is supported by conductivity measurements. These findings with DABCO and in the case of TEA at low concentrations are in full agreement with reactions (1)–(3) of electron transfer and the subsequent back electron transfer (4). On the other hand, for TEA at higher concentrations (>1 mM) a conservation of the charges within the longer lived intermediates occurs (Fig. 6). The conductivity signal decays by second-order kinetics (at least to a major extent), whereby the half-lives in the presence of 5 M water are longer with respect to neat acetonitrile. The nature of the radical derived by the second reduction step in dry and in wet acetonitrile can be specified as neutral radical since formation of the radical anion via (7b) would result in a conductivity change, in contrast to the findings in all cases. Thus, H-atom transfer (7a) is suggested as origin of the delayed formation of the nitronaphthalene radicals.
The signal decreases in the 1–100 ms range to a value which is still larger than the initial one and levels off within seconds, i.e. finally, release of OH− and H+ results in neutralization (Scheme 2). This is consistent with termination process (8) and OH− release. If a proton would be released prior to OH− elimination, a strong negative signal is expected in alkaline solution due to the removal of bulk OH−. On the other hand, OH− elimination without rapid neutralization would increase the signal markedly due to the larger equivalent conductivity of OH− (Λ
= 180 cm2
Ω−1 mol−1) with respect to that of an organic anion (Λ
≤ 50 cm2
Ω−1 mol−1).
The primary products from 1-NN, 2-NN and MNN at low conversion should be attributed to the nitrosonaphthalenes. In the photoreduction of nitrobenzenes by alcohols the nitroso compounds are postulated as labile intermediates, but thermal reactions lead to phenylhydroxylamine (ArNHOH) and further products, such as azoxybenzenes.9–11,30 An intermediate prior to phenylhydroxylamine could be the phenylnitroxide (phenylaminoxyl) radical (Ar–N˙–OH) which may be present in basic solution as nitrosobenzene radical anion.2,13 In the photoreduction of nitrobenzylidene malonic derivatives by TEA the nitroso compounds are also postulated as intermediates prior to photoproducts from secondary excitation.32
Upon UV irradiation of MNN in argon-saturated acetonitrile in the presence of TEA or DEA, formation of the corresponding naphthylhydroxylamine may become important. Once a considerable amount of nitrosonaphthalene is formed, a further reaction competing with (7) and yielding the naphthylhydroxylamine via the naphthylnitroxide (napthylaminoxyl) radical is proposed (Scheme 3).
| | |
Et2N–˙CHMe + ArNO → Et2NCHCH2
+ Ar–N˙–OH
| (12) |
| | |
2 × Ar–N˙–OH → ArNO + ArNHOH
| (13) |
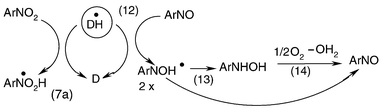 |
| | Scheme 3 | |
Reactions (12) and (13) are supported by the finding that addition of oxygen to the photoproduct of MNN leads to a spectrum with red-shifted maximum (curve 3 in Fig. 3b). Since the oxygen effect is not reversible (Fig. 3b), it is tentatively attributed to reaction (14), whereas a hypothetical reaction of oxygen with the nitrosonaphthalene has to be excluded.
| | |
ArNHOH + 1/2 O2
→ ArNO + H2O
| (14) |
The above measurements refer to deoyxgenated dry and wet acetonitrile solution, unless otherwise indicated. For the three nitronaphthalenes, due to the high kq values, reaction (3) could compete successfully with triplet quenching by oxygen, e.g. for donor concentrations of [TEA] > 0.5 mM in air-saturated acetonitrile. The important quenching reactions are, however, formation of the rather unreactive O2˙− by reaction with the Ar˙NO2H, ArNO2˙− and Et2N–˙CHMe type radicals.38 Therefore, Φd is very low when oxygen is present (Table 2). This is also in agreement with much shorter half-lives in the decay kinetics of the optical and conductometric signals in the presence of oxygen. Concerning different photoproducts than in the literature,25 the various possibilities for quenching by oxygen (even in trace amounts), apart from irradiation of the nitrosonaphthalenes, should not be ignored.
It is worth mentioning that formation of the products of 1- and 2-NN in neat argon-saturated propan-2-ol is rather inefficient; Φd is smaller than 0.01 and 0.04, respectively.29 Albeit the Ar˙NO2H radicals could be observed under these conditions (not shown), in agreement with H-atom abstraction from the alcohol and a shorter triplet lifetime (τT
= 5 and 3 μs, respectively) than in acetonitrile, their termination does not yield further transients absorbing above 360 nm. Virtually the same occurs in the presence of 5 M water, but a remaining transient absorbing at 400 nm appears, when the pH is shifted to 11–13. The latter is in agreement with the larger reactivity of the alcohol radical anion in the alkaline range with respect to its conjugate acid. The remaining transient absorbance is further enhanced (3–4 fold) on addition of 1–10 mM TEA. This is just the condition of excess OH− ions (apart from the solvent), used for the results presented in Figs. 2 and 4a. We therefore conclude that the nitronaphthalene–propan-2-ol system even at alkaline pH is less efficient in photoreduction than the nitronaphthalene–TEA system, which reveals rather large Φd values (Table 2). Moreover, the presence of an inert solvent is not required since triplet quenching by the amine successfully competes with H-atom abstraction from the alcohol.
Conclusion
The photoreduction by TEA of 1-methoxy-4-nitronaphthalene, 1- and 2-nitronaphthalene involves two subsequent steps: electron transfer from TEA to the lowest triplet state and H-atom transfer from the α-aminoalkyl radical to the substrate. These nitroaromatic radicals subsequently undergo a slow termination reaction when oxygen is excluded, yielding the corresponding nitrosonaphthalenes as labile products and finally products derived therefrom.
Acknowledgements
We thank Professor Wolfgang Lubitz for his support and Mr Leslie J. Currell and Horst Selbach for technical assistance.
References
- S. G. Cohen, A. Parola and G. H. Parsons, Jr., Chem. Rev., 1973, 73, 141 CrossRef CAS.
-
M. Chanon and
L. Eberson, in Photoinduced Electron Transfer, Eds. M. A. Fox and M. Chanon, part A, Elsevier, Amsterdam, 1988, p. 409 Search PubMed.
- J. C. Scaiano, J. Phys. Chem., 1981, 85, 2851 CrossRef CAS.
-
(a) I. Amada, M. Yamaji, S. Tsunoda and H. Shizuka, J. Photochem. Photobiol., A, 1996, 95, 27 Search PubMed;
(b) P. Aspari, N. Ghoneim, E. Haselbach, M. v. Raumer, P. Suppan and E. Vauthey, J. Chem. Soc., Faraday Trans., 1996, 92, 1689 RSC;
(c) R. Gonzales-Blanco, J. L. Bourdelande and J. Marquet, J. Org. Chem., 1997, 62, 6903 CrossRef CAS.
- A. Demeter, L. Biczók, T. Bérces, V. Wintgens, P. Valat and J. Kossanyi, J. Phys. Chem., 1993, 97, 3217 CrossRef CAS.
- M. Goez and I. Sartorius, J. Am. Chem. Soc., 1993, 115, 11123 CrossRef CAS.
- H. Görner, D. Döpp and A. Dittmann, J. Chem. Soc., Perkin Trans. 2, 2000, 1723 RSC.
- G. A. Davis, J. D. Gresser and P. A. Carapellucci, J. Am. Chem. Soc., 1971, 93, 2179 CrossRef CAS.
- J. A. Barltrop and N. J. Bunce, J. Chem. Soc. (C), 1968, 1467 RSC.
- R. Hurley and A. C. Testa, J. Am. Chem. Soc., 1968, 90, 1949 CAS.
- W. Trotter and A. C. Testa, J. Am. Chem. Soc., 1968, 90, 7044 CrossRef CAS.
- Y. Ogata, M. Tsuchida and Y. Takagi, J. Am. Chem. Soc., 1957, 79, 3397 CrossRef CAS.
- Y. Ogata, Y. Sawaki, J. Mibae and T. Morimoto, J. Am. Chem. Soc., 1964, 86, 3854 CrossRef CAS.
- G. A. Russell, E. J. Geels, F. J. Smentowski, K.-Y. Chang, J. Reynolds and G. Kaupp, J. Am. Chem. Soc., 1967, 89, 3821 CrossRef CAS.
- G. G. Wubbels, J. W. Jordan and N. S. Mills, J. Am. Chem. Soc., 1973, 95, 1281 CrossRef CAS.
- D. Döpp and D. Müller, Recl. Trav. Chim. Pays-Bas, 1979, 98, 297 Search PubMed.
- N. Levy and M. D. Cohen, J. Chem. Soc., Perkin Trans. 2, 1979, 553 RSC.
- P. Wan and K. Yates, J. Chem. Soc., Chem. Commun., 1981, 1023 RSC.
- J. Marquet, M. Moreno-Maňas, A. Vallribera, A. Virgili, J. Bertran, A. Gonzales-Lafont and J. M. Lluch, Tetrahedron, 1987, 43, 351 CrossRef CAS.
- S. Fukuzumi and Y. Tokuda, Bull. Chem. Soc. Jpn., 1992, 65, 831 CAS.
-
(a) R. L. Letsinger and R. R. Hautala, Tetrahedron Lett., 1969, 48, 4205 CrossRef;
(b) K. Obi, J. W. Bottenheim and I. Tanaka, Bull. Chem. Soc. Jpn., 1973, 46, 1060 CAS.
-
(a) E. Havinga and J. Cornelisse, Pure Appl. Chem., 1976, 47, 1 CAS;
(b) J. G. Lammers and J. Cornelisse, Isr. J. Chem., 1977, 16, 299 Search PubMed.
-
A. N. Frolov,
N. A. Kuznetsova,
V. A. El'tsov and
N. I. Rtishchev, Zh. Org. Khim., 1973, 9, 963
(J. Org. Chem., USSR (Engl Transl.), 1973, 9, 988) Search PubMed.
-
A. N. Frolov,
V. A. El'tsov,
I. M. Sosonkin and
N. A. Kuznetsova, Zh. Org. Khim., 1973, 9, 973 (J. Org. Chem., USSR (Engl Transl.), 1973, 9, 999) Search PubMed.
- N. J. Bunce, S. R. Cater, J. C. Scaiano and L. J. Johnston, J. Org. Chem., 1987, 52, 4214 CrossRef CAS.
-
(a) C. Capellos and G. Porter, J. Chem. Soc., Faraday Trans. 2, 1974, 1159 RSC;
(b) C. Capellos and K. Suryanarayanan, Int. J. Chem. Kinet., 1976, 8, 529 CrossRef CAS.
- L. J. A. Martins, M. Mendes, M. M. Fernandes, T. J. Kemp, S. J. Formosinho and J. S. Branco, J. Chem. Soc., Faraday Trans., 1991, 87, 3617 RSC.
-
J. Lin, Ph. D. Thesis, University of Duisburg, 1994 Search PubMed.
- D. Döpp, Top. Curr. Chem., 1975, 55, 51 Search PubMed.
-
D. Döpp, CRC Handbook of Photochemistry and Photobiology, Eds. W. M. Horspool and P.-S. Song, CRC Press, Boca Raton, 1995, p. 1019 Search PubMed.
-
T.-I. Ho and
Y. L. Chow, in The Chemistry of Amino, Nitroso, Nitro and Related Groups, part 2, Ed. S. Patai, Wiley, Chichester, 1996, p. 747 Search PubMed.
-
(a) F. M. Abd El Latif, M. A. Barsy, E. A. Rady, M. E. Hassan and M. A. El Maghraby, J. Photochem. Photobiol., A: Chem., 1999, 121, 111 Search PubMed;
(b) M. Fawi and M. Abd El Latif, J. Photochem. Photobiol., A: Chem., 2001, 141, 241 Search PubMed.
- P. Zuman and Z. Fijalek, J. Electroanal. Chem., 1990, 296, 583 CrossRef CAS.
-
(a) H. Görner, K.-D. Warzecha and M. Demuth, J. Phys. Chem. A, 1997, 101, 9964 CrossRef CAS;
(b) K.-D. Warzecha, H. Görner and M. Demuth, J. Chem. Soc., Faraday Trans., 1998, 94, 1701 RSC;
(c) C. Laurich, H. Görner and H. J. Kuhn, J. Photochem. Photobiol. A, 1998, 112, 29 Search PubMed;
(d) H. Görner and H. J. Kuhn, J. Chem. Soc., Perkin Trans. 2, 1999, 2671 RSC.
- H. G. Heller and J. R. Langan, J. Chem. Soc., Perkin Trans. 1, 1981, 341 Search PubMed.
- K.-D. Asmus, A. Wigger and A. Henglein, Ber. Bunsenges. Phys. Chem., 1966, 70, 862 Search PubMed.
- J. A. Barltrop and R. J. Owers, J. Chem. Soc., Chem. Comm., 1970, 1462 RSC.
- R. F. Anderson, W. A. Denny, W. Li, J. E. Packer, M. Tercel and W. R. Wilson, J. Phys. Chem. A, 1997, 101, 9704 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2002 |
Click here to see how this site uses Cookies. View our privacy policy here.