15N NMR spectra, tautomerism and diastereomerism of 4,5-dihydro-1H-1,2,3-triazoles
Klaus
Banert
*a,
Jens
Lehmann
a,
Helmut
Quast
b,
Georg
Meichsner
b,
Dieter
Regnat
b and
Bernhard
Seiferling
b
aInstitut für Chemie der Technischen Universität Chemnitz, Strasse der Nationen 62, D-09111 Chemnitz, Germany. E-mail: klaus.banert@chemie.tu-chemnitz.de; Fax: +49 371 531 1839; Tel: +49 371 531 1463
bInstitut für Organische Chemie der Universität Würzburg, Am Hubland, D-97074 Würzburg, Germany
First published on 3rd December 2001
Abstract
Despite the great number of 4,5-dihydro-1H-1,2,3-triazoles synthesized, 15N NMR data of these heterocycles are extremely rare. The aim of this paper is to present such data and examples of their application. The compounds investigated have been synthesized according to the given references or procedures. Their 15N NMR spectra were measured at natural abundance. For some compounds, the chemical shift assignments were confirmed with the help of 15N labelled material. The influences on 15N chemical shifts of substitution pattern, solvent and concentration were investigated. Additionally, some lanthanide induced shift (LIS) investigations were performed. 13C labelled compounds were employed as tools to provide the assignment of tautomeric structures.
Introduction
15N NMR spectroscopy is a powerful tool for the characterization of nitrogen containing compounds because 15N chemical shifts strongly depend on structural and electronic features which hence result in a broad shift range of approximately 900 ppm, i.e. −400 to +500 ppm relative to nitromethane as external reference.1–3 Despite and sometimes through the influence of solvents as well as concentration, pH value and temperature of the solutions on 15N shifts, many problems, including for example tautomerism2–4 or positions of protonation of heterocycles,3,5 could be solved with the help of 15N NMR spectroscopy.The number (more than 3300) of 4,5-dihydro-1H-1,2,3-triazoles that are registered by Chemical Abstracts attests to the importance of this class of heterocycles for many areas ranging from organic syntheses to pharmaceutical research.6 Nevertheless, 15N NMR data of these heterocyclic compounds are surprisingly rare. To the best of our knowledge, only a handful have been published in previous papers from our group.7–9 The primary goal of the present study is to fill this gap and to utilize such data for the elucidation of the structures of tautomers and diastereomers.
The dihydro-1,2,3-triazoles investigated have been synthesized according to procedures reported in the literature or described in the Experimental section. In most cases, they were prepared by 1,3 dipolar cycloaddition of an azide to an alkene. Their 15N NMR spectra were measured at natural abundance (0.37% 15N) except for some cases (compare Fig. 1) in which the shift assignment had to be confirmed with the help of 15N labelling.
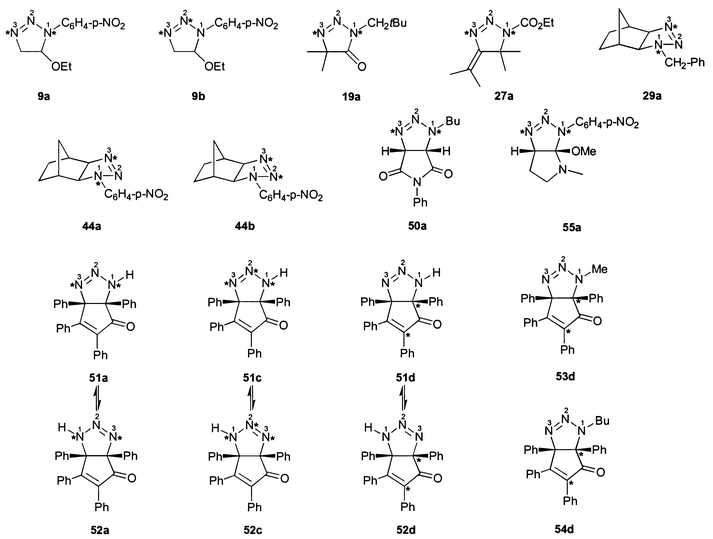 | ||
| Fig. 1 15N and 13C labelled compounds. | ||
Experimental
The known dihydrotriazoles 1–32, 34, 36–38, 40, 41, 44, 46–53, 55–60 and 62 were prepared according to the procedures given in the references listed in Table 3. The compounds 33, 35, 39, 42, 43 and 45 were obtained by 1,3 dipolar cycloaddition of the corresponding azides to norbornene (molar ratio azide ∶ norbornene ca. 1 ∶ 2) under the reaction conditions specified in Table 1. The physical and spectroscopic data of these new compounds are summarized in Table 2. Chemical shifts are given in ppm and J values are given in Hz.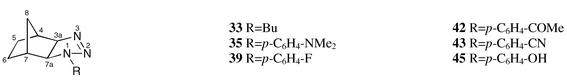
| Compound | 33 | 35 | 39 | 42 | 43 | 45 |
|---|---|---|---|---|---|---|
| a May be exchanged. | ||||||
| δ 1H (ppm) | (in CDCl3) | (in CDCl3) | (in CDCl3) | (in CDCl3) | (in CDCl3) | (in DMSO-d6) |
| 3a-H | 4.34 (d, J 10) | 4.52 (d, J 9.4) | 4.57 (d, J 9.7) | 4.64 (d, J 9) | 4.67 (d, J 8.8) | 4.51 (d, J 9.4) |
| 4-H | 2.61a (broad s) | 2.62 (broad s) | 2.61 (broad s) | 2.65 (broad s) | 2.63 (broad s) | |
| 5-H/6-H | 1.15–1.70 (m) | 1.20–1.85 (m) | 1.20–1.80 (m) | 1.20–1.80 (m) | 0.9–1.9 (m) | |
| 7-H | 2.30a (broad s) | 2.74 (broad s) | 2.78 (broad s) | 2.80 (broad s) | 2.85 (broad s) | |
| 7a-H | 3.19 (d, J 10) | 3.72 (d, J 9.4) | 3.69 (d, J 9.7) | 3.73 (d, J 9) | 3.70 (d, J 8.8) | 3.78 (d, J 9.4) |
| 8-H | 1.12 (s) | 1.16 (s) | 1.15 (s) | 1.15 (s) | [in 0.9–1.9 (m)] | |
| Other hydrogens | 0.93 (t, 4′-H) | 6.79 (m, arom.) | 6.80–7.40 | 7.30 (m, arom.) | 7.32 (m, arom.) | 6.75 (m, arom.) |
| 3.53 (t, 1′-H) | 7.23 (m, arom.) | (m, arom.) | 7.95 (m, arom.) | 7.62 (m, arom.) | 7.10 (m, arom.) | |
| 1.15–1.70 | ||||||
| (m, 2′-H/3′-H) | ||||||
| δ 13C (ppm) | (in CDCl3) | (in CDCl3) | (in CDCl3) | (in CDCl3) | (in CDCl3) | (in DMSO-d6) |
| C-3a | 85.3 | 84.7 | 85.9 | 86.4 | 86.9 | 84.8 |
| C-4/C-7 | 40.8, 41.3 | 39.6, 40.6, 40.8 (C-1′) | 39.5, 40.9 | 39.2, 40.4 | 39.5, 40.5 | |
| C-5/C-6 | 24.8, 25.7 | 24.3, 24.9 | 24.4, 25.0 | 24.2, 24.8 | 24.2, 24.9 | 24.1, 24.8 |
| C-7a | 62.8 | 60.6 | 60.1 | 59.0 | 58.9 | 60.1 |
| C-8 | 30.7, 32.3 (C-2′) | 31.5 | 31.7 | 31.4 | 31.5 | |
| Other carbons | 13.7 (C-4′) 20.0 (C-3′) 48.0 (C-1′) | 113.4, 115.1, 130.9, 146.2 (arom.) | 114.7, 115.6, 136.4, 157.8 (arom.) | 25.6 (COMe) 112.5, 129.6, 130.1, 143.3 (arom.), 195.5 (CO) | 103.5 (CN) 113.4, 118.7, 133.1, 143.0 (arom.) | 115.4, 115.8, 132.5, 152.5 (arom.) |
| Melting point /°C | (colorless oil) | 123 (ether–petroleum ether) | 100.5–101 (ether–petroleum ether) | 144–145 | 140–140.5 (petroleum ether–ether–methanol) | 156–160 (THF) |
| Analysis | ||||||
| Formula | C11H19N3 | C15H20N4 | C13H14FN3 | C15H17N3O | C14H14N4 | C13H15N3O |
| Found (calc.) (%) | ||||||
| C | 68.19 (68.35) | 70.15 (70.28) | 67.56 (67.51) | 70.39 (70.57) | 70.64 (70.57) | 67.84 (68.10) |
| H | 9.86 (9.91) | 7.88 (7.80) | 6.10 (6.10) | 6.70 (6.71) | 5.93 (5.92) | 6.65 (6.59) |
| N | 22.00 (21.74) | 21.90 (21.86) | 18.20 (18.17) | 16.30 (16.46) | 23.60 (23.51) | 18.20 (18.33) |
| IR/cm−1 | (in CCl4) | (in CCl4) | (in CCl4) | (in CCl4) | (in CCl4) | (KBr) |
| 1450, 1460, 1470, 2880, 2960 | 1095, 1260, 1330, 1475, 1510, 2960 | 1225, 1490, 1505, 2960 | 1265, 1365, 1495, 1595, 1675 (CO), 2960 | 1370, 1495, 1510, 1600, 2220 (CN), 2960 | 1125, 1140, 1230, 1360, 1440, 1460, 1510, 2960 | |
| Compound | 15N Chemical shifts | Reference for preparation | |||
|---|---|---|---|---|---|
| N-1 | N-2 | N-3 | Other nitrogen | ||
| a In CD3OD. b The chemical shifts have been reported in ref. 7. c The chemical shifts have been reported in ref. 8. d In DMSO-d6. e In DMSO-d6 at 363 K. f Doublet with 1JNH = 100.5 Hz. g No assignment of meso and rac compounds, the chemical shifts have been reported in ref. 9. | |||||
| 1 | −195.5 | 54.6 | −31.4 | 22, 23 | |
| 2 | −194.6 | 53.3 | −28.4 | 22, 24 | |
| 3 | −182.8 | 35.0 | −29.3 | 25 | |
| 4 | −182.7 | 33.3 | −42.0 | 26 | |
| 5 | −174.4 | 35.7 | −34.4 | 27 | |
| 6 | −178.8 | 34.7 | −29.9 | 28 | |
| 7 | −195.5 | 52.8 | −56.2 | −127.1 (CN) | 29 |
| 8 | −175.4 | 34.6 | −30.4 | 28 | |
| 9 | −168.3 | 32.7 | −17.6 | −12.7 (NO2) | 30, 31 |
| 10 | −182.5 | 37.6 | −50.7 | −286.4 (NEt2) | 32 |
| 11 | −182.6 | 34.1 | −46.8 | 26 | |
| 12 | −177.1 | 36.3 | −48.4 | 33 | |
| 13 | −176.9 | 35.8 | −46.8 | 33 | |
| 14 | −177.6 | 44.4 | −5.3 | 34 | |
| 15 | −179.5 | 43.5 | −8.2 | 34 | |
| 16 | −172.5 | 52.4 | −12.1 | 35 | |
| 17 | −172.4 | 52.0 | −10.4 | 35 | |
| 18 | −148.4 | 43.7 | 32.2 | 36 | |
| 19 | −163.1 | 44.0 | 32.8 | 36 | |
| 20 a , b | −169.1 | 39.6 | 30.4 | 37 | |
| 21 b | −173.1 | 42.8 | 27.4 | 37 | |
| 22 | −179.7 | 32.4 | −6.9 | 35 | |
| 23 | −172.9 | 36.4 | 3.5 | −160.4 (NMe) | 38 |
| 24 | −183.1 | 34.6 | 5.4 | −161.2 (NMe) | 38 |
| 25 c | −140.2 | 36.5 | −18.8 | 8 | |
| 26 | −167.7 | 18.3 | −36.3 | 39 | |
| 27 | −159.3 | 12.4 | −11.4 | 39 | |
| 28 | −184.0 | 48.0 | −25.8 | 40 | |
| 29 | −188.3 | 47.6 | −36.1 | 41 | |
| 30 | −166.9 | 47.0 | −36.5 | 42 | |
| 31 | −201.2 | 38.2 | −17.4 | 43 | |
| 32 | −162.9 | 33.3 | 2.5 | 44 | |
| 33 | −190.1 | 47.9 | −39.0 | see Exp. | |
| 34 | −168.7 | 38.5 | −25.4 | 45 | |
| 35 | −176.9 | 33.5 | −35.4 | −339.5 (NMe2) | see Exp. |
| 36 | −176.5 | 33.5 | −31.2 | 46 | |
| 37 | −175.0 | 33.5 | −29.3 | 46 | |
| 38 | −173.9 | 33.5 | −27.5 | 41, 46, 47 | |
| 39 | −176.0 | 33.1 | −26.7 | see Exp. | |
| 40 | −174.9 | 32.6 | −24.0 | 41 | |
| 41 | −175.0 | 32.2 | −23.8 | 44, 46 | |
| 42 | −170.9 | 32.2 | −17.0 | see Exp. | |
| 43 | −170.8 | 31.6 | −14.5 | −127.5 (CN) | see Exp. |
| 44 | −169.7 | 31.7 | −12.1 | −12.5 (NO2) | 41, 46 |
| 45 d | −175.7 | 33.9 | −28.7 | see Exp. | |
| 46 | −172.6 | 33.8 | −26.8 | −324.7 (pyrrolidino) | 48 |
| 47 | −137.9 | 38.1 | 5.6 | −318.1 (pyrrolidino) | 31 |
| 48 | −175.9 | 33.9 | −23.7 | −329.3 (morpholino) | 48, 49 |
| 49 d | −178.5 | 34.2 | −37.1 | −197.6 (imide-N) | 44, 50 |
| 50 | −193.7 | 46.2 | −53.6 | −198.3 (imide-N) | 44 |
| 51 e | −189.4f | 34.6 | −40.9 | 19 | |
| 52 e | −193.4f | 37.0 | −37.6 | (19) | |
| 53 | −200.1 | 36.0 | −46.2 | 19 | |
| 54 | −183.2 | 40.0 | −41.9 | see Exp. | |
| 55 | −170.0 | 41.4 | −11.9 | −12.4 (NO2) | 51 |
| −321.0 (NMe) | |||||
| 56 | −172.1 | 34.3 | −15.9 | 28 | |
| 57 | −168.9 | 35.0 | −12.0 | 52 | |
| 58 | −159.4 | 62.3 | −12.1 | 53 | |
| 59 | −161.3 | 58.4 | −5.4 | 53 | |
| 60 | −152.7 | 59.4 | −11.2 | 53 | |
| 61 | −194.1 | 51.0 | −32.9 | see Exp. | |
| 62 g | −170.5 | 36.5 | −26.8 | 9 | |
| −170.5 | 36.5 | −27.3 | |||
Compound 54: Butyl azide10 (600 mg, solution in Et2O, 64%, 3.88 mmol) and 2,3,4,5-tetraphenylcyclopentadienone (850 mg, 2.21 mmol) were dissolved in benzene (10 ml). The mixture was sealed in a glass tube and heated to 75 °C for 14 days. After filtration, the solvent and remaining BuN3 were removed in vacuo at 45 °C. The residue was treated with ethanol. After separation of the insoluble starting material, the solvent was evaporated to give 610 mg (1.26 mmol, 57%) of pure 54 as yellow crystals, mp 60–61 °C (from EtOH) (Found: C, 82.12; H, 6.21. Calc. for C33H29N3O: C, 81.96; H, 6.04%); νmax (CCl4)/cm−1 1708 (CO); δH(300 MHz; CDCl3) 0.86 (3 H, t, 4′-Me), 1.35 (2 H, sext., 3′-H), 1.81 (2 H, quint., 2′-H), 3.43 (1 H, m, 1′-H), 3.69 (1 H, m, 1′-H), 6.8–7.6 (20 H, m, arom. H); δC(75 MHz, CDCl3) 13.7 (C-4′), 20.1 (C-3′), 31.1 (C-2′), 47.1 (C-1′), 77.3 (C-5), 98.7 (C-4), 127.1, 127.5, 127.7, 127.9, 128.4, 129.5, 129.6, 130.6, 131.1, 133.0, 133.1, 135.8 (arom. C), 141.5 (C-7), 167.9 (C-8), 199.5 (C-6).
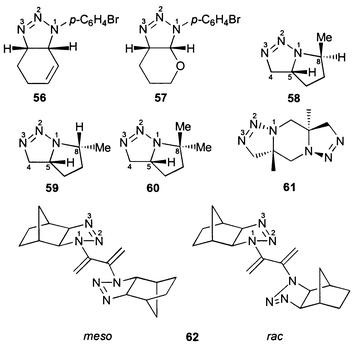
Compound 61 (with a structure assumed to be analogous to that of allyl azide dimer16): A solution of methallyl azide17 (5.0 g, 51.5 mmol) in toluene (50 ml) was heated under reflux for 3 days. The solvent was removed in vacuo to yield the crude product (2.1 g, 42%) which furnished a colorless solid (0.64 g, 13%), mp 220 °C (from CH2Cl2–Et2O); δH(300 MHz, CDCl3) 1.40 (3 H, s, Me), 3.37 (1 H, d, J 14.4, 6-H), 3.75 (1 H, d, J 14.4, 6-H), 3.68 (1 H, d, J 16.3, 4-H), 4.40 (1 H, d, J 16.3, 4-H); δC(75 MHz, CDCl3) 19.6 (Me), 47.0 (C-6), 58.9 (C-5), 74.9 (C-4).
Compounds 9a, 19a, 27a, 29a, 44a, 50a and 55a, 15N labelled in positions 1 and 3 (compare Fig. 1), were synthesized from the corresponding 15N labelled azides. These were obtained by nucleophilic substitution reactions from sodium azide having both terminal positions enriched with 15N (49%). 15N labelled neopentyl azide was synthesized in 97% yield from 1-iodo-2,2-dimethylpropane and molten (n-C16H33)Bu3P+15N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N15N−, which was prepared from 15N labelled sodium azide according to
the modified method reported in ref. 18. The heterocycles 51a/52a and 51c/52c were obtained from appropriately labelled sodium azide according to the procedure described in ref. 19. Compounds 9b and 44b, 15N enriched at positions 2 and 3, were obtained by a 1,3 dipolar cycloaddition reaction from p-nitrophenyl azide, which was prepared by coupling of p-nitrobenzenediazonium chloride with sodium azide, having both terminal positions labelled with 15N (49%). The resulting p-nitrophenyl azide was labelled weakly in the β and strongly in the γ position. The 13C
labelled compounds 51d/52d, 53d were prepared according to ref. 19 and 54d as described above for 54, respectively, using 2,3,4,5-tetraphenylcyclopentadienone labelled with 10%
13C in the positions 2 and 5. This starting material was prepared as described20 from 1,2-diphenylethanedione and 1,3-13C-1,3-diphenylpropan-2-one. The latter was obtained from 2-13C-2-phenylacetic acid (10%
13C by mixing of 13C labelled and unlabelled material in a 1 ∶ 9 ratio).21 The positions of 13C labelling of all substances were checked by 13C NMR spectroscopy.
N15N−, which was prepared from 15N labelled sodium azide according to
the modified method reported in ref. 18. The heterocycles 51a/52a and 51c/52c were obtained from appropriately labelled sodium azide according to the procedure described in ref. 19. Compounds 9b and 44b, 15N enriched at positions 2 and 3, were obtained by a 1,3 dipolar cycloaddition reaction from p-nitrophenyl azide, which was prepared by coupling of p-nitrobenzenediazonium chloride with sodium azide, having both terminal positions labelled with 15N (49%). The resulting p-nitrophenyl azide was labelled weakly in the β and strongly in the γ position. The 13C
labelled compounds 51d/52d, 53d were prepared according to ref. 19 and 54d as described above for 54, respectively, using 2,3,4,5-tetraphenylcyclopentadienone labelled with 10%
13C in the positions 2 and 5. This starting material was prepared as described20 from 1,2-diphenylethanedione and 1,3-13C-1,3-diphenylpropan-2-one. The latter was obtained from 2-13C-2-phenylacetic acid (10%
13C by mixing of 13C labelled and unlabelled material in a 1 ∶ 9 ratio).21 The positions of 13C labelling of all substances were checked by 13C NMR spectroscopy.
NMR spectra were recorded from 0.5 to 1.5 molar solutions in CDCl3 (unless stated otherwise) at a temperature of 309 K (unless stated otherwise) using standard pulse programs. 15N NMR spectra were recorded without proton decoupling on BRUKER WH-400 and AMX-400 spectrometers operating at 40.53 MHz and on a VARIAN Gemini 2000 spectrometer operating at 30.40 MHz. Nitromethane was employed as external reference (δ = 0 ppm) without susceptibility correction using an inner capillary tube of the standard. 14N NMR spectra were recorded on a BRUKER WH-400 spectrometer operating at 28.89 MHz. 1H NMR spectra were recorded on BRUKER WH-400 and AMX-400 spectrometers operating at 400 MHz and on a VARIAN Gemini 2000 spectrometer operating at 300 MHz and referenced with TMS or solvent signals, which were standardized to TMS. 13C NMR spectra were recorded on BRUKER WH-400 and AMX-400 spectrometers operating at 100.58 MHz and on a VARIAN Gemini 2000 spectrometer operating at 75.43 MHz. Referencing was performed as described for 1H NMR spectra.
Results and discussion
15N NMR chemical shifts and assignments of signals
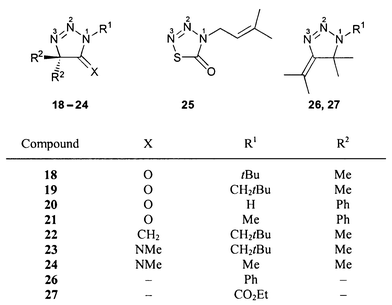
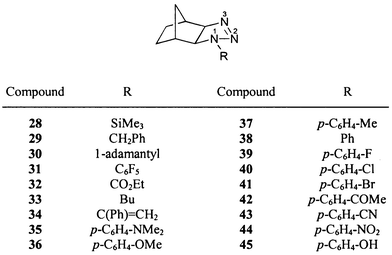
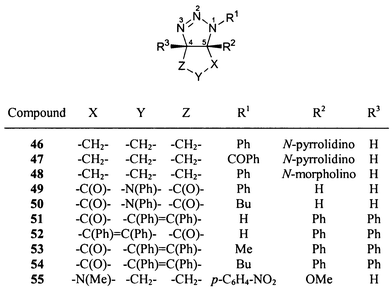
The 15N NMR chemical shifts of all dihydrotriazoles studied (1–62, Schemes 1–5) are summarized in Table 3. Although the 15N NMR spectra were measured without proton decoupling, no long-range JNH couplings could be resolved. The numbering scheme of the molecules does not follow in all cases the nomenclature rules for the sake of the comparability of chemical shifts, i.e. N-1 is always the highly substituted, amine-like (saturated) nitrogen atom of the dihydrotriazole ring bonded to N-2 and C-5, see for example 18–21. The sulfur heterocycle 25 [1,2,3,4-thiatriazol-5(4H)-one] could be regarded as a formal derivative of a 4,5-dihydro-1H-1,2,3-triazole. There were no differences in 15N chemical shifts observed for the 15N labelled (Fig. 1) and the unlabelled compounds.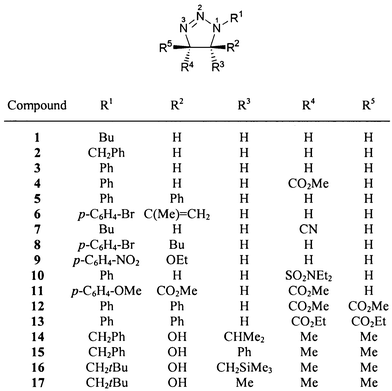 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
 | ||
| Scheme 4 | ||
 | ||
| Scheme 5 | ||
For the assignment of the 15N NMR signals of the 4,5-dihydro-1H-1,2,3-triazoles to N-1, N-2 and N-3, the electronic difference between pseudo-sp3 and sp2 hybridized nitrogen atoms was used to assign the signal at highest field to N-1. For the unequivocal distinction of N-2 and N-3 (both sp2 hybridized nitrogen atoms), 29a was investigated, which is labelled at the N-1 and N-3 positions.
The amine-like nitrogen N-1 shows in all cases the signal at the highest field in the range δ = −201 to −138 ppm. The resonance of nitrogen N-2 is observed usually at lowest field, i.e. in the range δ = 12 to 62 ppm. The chemical shift of nitrogen N-3 is found in the range −54 to 6 ppm, except in the case of compounds with a ring carbonyl group (18–21) which causes strong downfield shifts to δ values of ca. 30 ppm.
Generally, the 15N NMR data of 4,5-dihydro-1H-1,2,3-triazoles are clearly distinguished from those of cyclic azimines (4,5-dihydro-1H-1,2,3-triazol-2-ium-1-ides) as structurally isomeric compounds54 and those of open chain triazenes55 as well.
The four 15N NMR resonances of 9 could be assigned by means of labelling N-1 and N-3 (9a) and N-2 and N-3 (9b), respectively (Fig. 1). Thus, the 15N NMR signals of N-3 (−17.6 ppm) and NO2 (−12.7 ppm) became assignable. For the compounds 44 (labelled 44a and 44b) and 55 (labelled 55a), a similar assignment was done.
For the 15N chemical shift assignment of dihydrotriazolone 19, especially to N-2 and N-3, the corresponding N-1 and N-3 15N labelled isotopomer 19a was prepared. The signal at δ = 32.8 ppm stems from N-3 without doubt; nitrogen N-2 gives rise to the remaining 15N resonance at δ = 44.0 ppm.
The assignments of the 15N NMR signals of the thiatriazolone 258 are easily performed by comparison with the data of 18–21.
The four nitrogen NMR signals of compound 50, especially the differentiation of N-1 and the imide-N, could be achieved by 15N labelling of N-1 and N-3 (compound 50a, Fig. 1).
Tautomeric and diastereomeric compounds
Several years ago, two of us employed 15N NMR spectroscopy for the elucidation of the prevailing tautomer of 5,5-diphenyldihydro-1,2,3-triazol-4(4H)-one, which exhibits the structure 20.7The bicyclic [3 + 2] cycloadduct obtained from tetraphenylcyclopentadienone and sodium azide under acidic conditions has been reported to have a single tautomeric structure, namely 51, without experimental evidence.19 However, our 15N spectrum of this substance in DMSO-d6 shows two signal sets with different intensity (ratio ca. 4 ∶ 1) in accord with the possible tautomers 51 and 52 (Fig. 1, Scheme 4, Table 3). The 13C NMR and 1H NMR spectra also indicate the presence of both tautomeric forms. To assign the tautomers, the 13C enriched compounds 51d/52d were investigated. 13C NMR spectroscopy proved that 51 is the major and 52 the minor tautomeric structure. By 13C labelling (53d), it could be evidenced that the methylation product of 51/52, proposed to be 53,19 really possesses this structure, which is homologous to that of 51. The other conceivable methylation product with a structure similar to that of 52 was not observed. The 13C chemical shifts of the bridgehead carbons (C-4, C-5) and the carbonyl groups of 51–53 and of the related compound 54 are summarized in Table 4.
| Carbon | 51 | 52 | 53 | 54 |
|---|---|---|---|---|
| In the case of the labelled compounds, signal enhancements were observed for C-4 of 52d and for C-5 of 51d, 53d and 54d. | ||||
| CO |
200.8 | 198.8 | 199.0 | 199.5 |
| C-4 | 97.7 | 97.2 | 99.0 | 98.7 |
| C-5 | 75.1 | 77.2 | 76.6 | 77.3 |
The intramolecular 1,3 dipolar cycloaddition reaction of 5-azidohex-1-ene did not afford a single diastereomer as reported in ref. 53; instead, a 4 ∶ 1 mixture of the exo (58) and the endo diastereomer (59) was formed (Scheme 5). The structure assignment was performed both by lanthanide induced shift experiments (Fig. 2) and by 2D NOESY NMR spectroscopy.
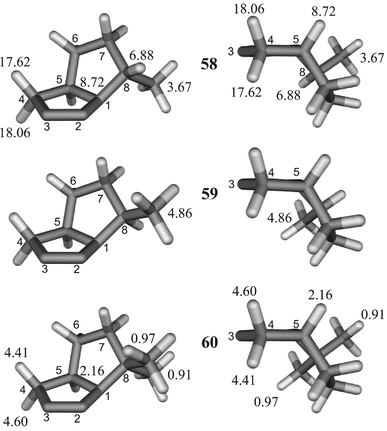 | ||
| Fig. 2 Effects of Yb(thd)3 shift reagent on 1H chemical shifts in Δδ (in ppm, downfield) at 1 ∶ 1 molar ratio (calculated by linear regression with a correlation coefficient better than 0.97) of selected protons of 58–60 (concentrations during LIS measurement: 58/59ca. 45 mmol l−1, 60ca. 3 mmol l−1). | ||
As proved by 15N NMR LIS experiments (see below) and by strong downfield shifts of the adjacent protons 4-H, the complexing site of 4,5-dihydro-1H-1,2,3-triazoles is N-3. Thus, the lanthanide induced shift of the signal of the methyl group is less effective in the case of 58 since the exo-methyl group is more distant from N-3 as compared to the case of 59. Additionally, the endo-proton geminal to the exo-methyl group of 58 (8-H) shows nearly the same LIS as the bridgehead proton (5-H). So a second piece of evidence for the exo position of the methyl group of the major isomer 58 is given. By adding the shift reagent to 60, the same influence on proton chemical shifts was observed although the effect was not as strong as for 58 and 59 (compare Fig. 2), probably caused by a smaller concentration of 60 during the measurement. The 2D NOESY spectrum shows cross signals between the methyl group and 5-H for 58 which indicates also the exo position of the methyl group. If the methyl group were in the endo position, a NOE should be observed between 5-H and 8-H which was not detected, however. The 1H and 13C NMR data of the 1,2,3-triazabicyclo[3.3.0]oct-2-enes 58–60 are summarized in Table 5.
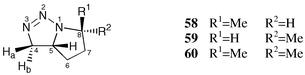
| 1H | 13C | |||
|---|---|---|---|---|
| a Signals of 59 partially covered by those of 58. | ||||
| 58 | 6-H (m) | 1.17 | Me | 21.7 |
| Me (d, J 6.9) | 1.33 | C-6 | 30.0 | |
| 7-H (m) | 1.41 | C-7 | 31.7 | |
| 6-H (m) | 1.81 | C-5 | 56.8 | |
| 7-H (m) | 1.84 | C-8 | 57.5 | |
| 5-H (m) | 3.66 | C-4 | 71.8 | |
| 4-Ha (dd, J 16.5, 9.6) | 4.07 | |||
| 4-Hb (dd, J 16.5, 2.6) | 4.29 | |||
| 8-H (m) | 4.34 | |||
| 59 a | Me (d, J 6.7) | 1.66 | Me | 16.8 |
| 5-H (m) | 3.60 | C-6/C-7 | 30.3 | |
| 4-Ha (dd, J 16.5, 9.7) | 4.13 | 31.8 | ||
| C-5/C-8 | 57.0 | |||
| 59.9 | ||||
| C-4 | 73.7 | |||
| 60 | exo-Me (s) | 1.28 | exo-Me | 25.9 |
| 6-H (m) | 1.30 | endo-Me | 25.6 | |
| 7-H (m) | 1.55 | C-6 | 30.0 | |
| endo-Me (s) | 1.65 | C-7 | 37.4 | |
| 6-H (m) | 1.86 | C-5 | 56.2 | |
| 7-H (m) | 1.86 | C-8 | 64.9 | |
| 5-H (m) | 3.76 | C-4 | 73.0 | |
| 4-Ha (dd, J 16.5, 9.9) | 4.10 | |||
| 4-Hb (dd, J 16.5, 2.7) | 4.32 | |||
Influence of concentration, solvents and shift reagents
The effect Δδ on the 15N chemical shifts caused by different concentrations of 4,5-dihydro-1H-triazoles was less than 2 ppm. For example, on comparison of a 0.64 molar and a 1.1 molar solution of 15 in CDCl3, the absolute Δδ values were as follows: 0.05 ppm for N-1, 0.39 ppm for N-2 and 1.64 ppm for N-3. The use of solvents other than CDCl3 (see Table 6) affects the signals of N-1 and N-2 (Δδ < 2 ppm) less than that of N-3 whose shift values vary up to 15 ppm (for 1 in CD3OD vs. cyclohexane-d12).| Compound | Solvent | 15N Chemical shifts | ||
|---|---|---|---|---|
| N-1 | N-2 | N-3 | ||
| 1 | CDCl3 | −195.5 | 54.6 | −31.4 |
| Cyclohexane-d12 | −195.9 | 56.4 | −24.4 | |
| CD3OD | −195.1 | 54.6 | −39.1 | |
| 28 | CDCl3 | −184.0 | 48.0 | −25.4 |
| Acetone-d6 | −184.1 | 48.0 | −21.2 | |
| Benzene-d6 | −184.8 | 47.7 | −20.9 | |
| DMSO-d6 | −183.7 | 48.0 | −22.4 | |
| 50 | CDCl3 | −193.7 | 46.2 | −53.6 |
| DMSO-d6 | −193.1 | 47.1 | −48.7 | |
The application of lanthanide induced shift reagents to 4,5-dihydro-1H-triazoles can cause strong changes of 15N shifts (Δδ > 150 ppm). Interaction of Yb(thd)3 [tris(2,2,6,6-tetramethylheptane-3,5-dionato)ytterbium(III)] and 38 leads to the strongest downfield shift for N-3 while the effect on N-1 was the smallest. This behavior was found with all investigated compounds. Its accordance with the calculated preference for protonation56 at N-3 allows the conclusion that this is the nitrogen which interacts with the shift reagent. The results are summarized in Fig. 3.
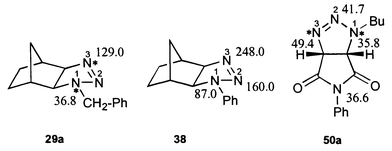 | ||
| Fig. 3 Effects of Yb(thd)3 shift reagent on 15N chemical shifts (Δδ in ppm, downfield) at 1 ∶ 1 molar ratio calculated by linear regression with a correlation coefficient better than 0.989. | ||
For 50a, not so strong downfield shifts were observed. This is probably caused by the additional two complexing sites (CO groups) in the molecule.
14N NMR measurements
Despite considerable line broadening, the 14N NMR spectrum of 1 (Fig. 4) consists of singlets which are sharp enough for the measurement of accurate chemical shifts. But this example is, unfortunately, not representative. In most other cases, no 14N NMR chemical shifts could be measured because of excessive broad lines.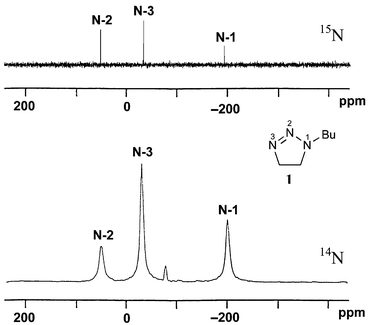
The effect of substituents on the 15N chemical shifts
Formal change of the N-1 substituent from aliphatic to aromatic causes strong and variable downfield shifts of the N-1 and N-3 signals, respectively, and a strong upfield shift of the N-2 resonance, see compounds 1 (Bu) and 2 (CH2Ph) vs.3 (Ph, Scheme 1) and 33 (Bu) and 29 (CH2Ph) vs.38 (Ph, Scheme 3) as well as 50 (Bu) vs.49 (Ph, Scheme 4). The effect on the N-1 shielding is similar in the case of amines, compare, for example, butylamine (δ = −359.4 ppm) with aniline (δ = −320.3 ppm).58 Furthermore, electronic (resonance) effects cause changes of the N-2 and N-3 chemical shifts.
If the N-1 substituent is changed from a phenyl to a carbonyl group, strong or even very strong downfield shifts of N-1 and N-3 signals are observed, compare 26 (Ph) vs.27 (CO2Et, Scheme 2) and 38 (Ph) vs.32 (CO2Et, Scheme 3) as well as 46 (Ph) vs.47 (COPh, Scheme 4).
A change from alkyl to silyl substituent at N-1 causes downfield shifts both of N-1 and N-3 signals, see 33 (Bu) vs.28 (SiMe3, Scheme 3). The fact that this shift is towards lower field is remarkable because in the case of amines the same structural change results in a shift into the opposite direction, see, for example, Bu–NMe2 (δ = −352.8 ppm)57 and Me3Si–NMe2 (δ = −378.5 ppm).58
As may be anticipated on consideration of the substituent effects discussed above, the downfield shifts of the signals of N-1 and N-3, and the opposite shifts of N-2, which originate from increasingly electron-withdrawing substituents at the para position of the phenyl rings of 35–44, result in satisfactory linear relationships with Hammett's σp constants59 (Fig. 5). While the signs of the slopes are as expected, their absolute values surprise, however. The signal of N-3 rather than that of N-1, to which the p-substituted phenyl ring is actually attached, is affected most strongly by variation of the p-substituent. On the other hand, the signal of N-2 is almost invariant toward these changes.
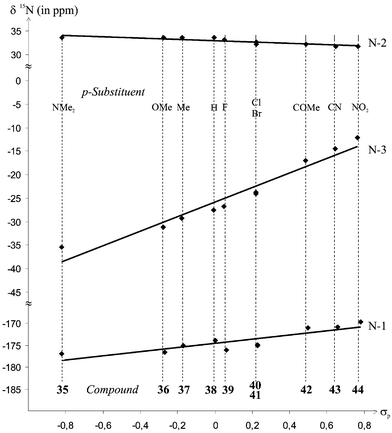 | ||
| Fig. 5 Correlation of Hammett σp constants vs.15N chemical shifts of compounds 35–44. | ||
Since 19F chemical shifts of p-substituted fluorobenzenes60 are linearly correlated with the σp constants of the substituents, it comes as no surprise that the 15N shifts of the 1-aryldihydrotriazoles 35–44 yield linear relationships with those 19F shifts as well (Fig. 6). Their slopes, viz. 0.302 for N-1, −0.078 for N-2 and 0.877 for N-3, show that the electronic effects of p-substituents on the 15N nuclei of 35–44 indeed closely resemble those on the 19F nuclei in the corresponding fluorobenzenes.
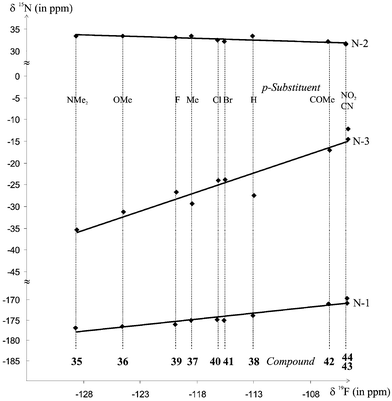 | ||
| Fig. 6 Correlation of 19F chemical shifts of p-substituted fluorobenzenes vs.15N chemical shifts of compounds 35–44. | ||
The butyl group at C-5 of 8 has a γ effect on N-1 while the C-5 isopropenyl group of 6 causes two γ effects on the same nitrogen. Thus, a small overall upfield shift is observed in the latter case.
Upfield shifts of the N-1 and N-2 resonances and very strong upfield shifts for the N-3 resonance are observed when the oxo group in position 5 is formally exchanged for an N-methylimino or a methylene group [compare 19vs.23vs.22 (Scheme 2)]. These upfield shifts are readily interpreted in terms of the electronegativity of the elements that are doubly bonded at C-5. However, the differences between the oxo compound (19) and the imino compound (23) are surprisingly greater than that between the latter (23) and the methylene compound (22).
Conclusions
15N NMR spectroscopy is still not widely used, and the presented chemical shift data of 4,5-dihydro-1H-triazoles and their dependence on the nature of substituents detailed here may be useful for further structure determinations of such heterocycles by this method or for the assignment of 15N signals of similar heterocyclic compounds.Acknowledgements
This work is dedicated to Professor Siegfried Hünig on the occasion of his 80th birthday. We thank Professor Dr H. Günther and his coworkers, especially Dr D. Moskau, for stimulating and helpful discussions while a great part of the measurements were performed at the University of Siegen. This research was supported by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie. K. B. and J. L. wish to thank Dynamit Nobel GmbH, Leverkusen, for providing chemicals.References
- S. Berger, S. Braun and H.-O. Kalinowski, NMR-Spektroskopie von Nichtmetallen, Band 2, 15N-NMR-Spektroskopie, Georg Thieme, Stuttgart, 1992 Search PubMed; M. Witanowski, L. Stefaniak and G. A. Webb, Annu. Rep. NMR Spectrosc., 1972, 5A, 395–464 Search PubMed; M. Witanowski, L. Stefaniak and G. A. Webb, Annu. Rep. NMR Spectrosc., 1977, 7, 117–244 Search PubMed; M. Witanowski, L. Stefaniak and G. A. Webb, Annu. Rep. NMR Spectrosc., 1981, 11B, 1–502 Search PubMed; M. Witanowski, L. Stefaniak and G. A. Webb, Annu. Rep. NMR Spectrosc., 1986, 18, 1–761 Search PubMed; M. Witanowski, L. Stefaniak and G. A. Webb, Annu. Rep. NMR Spectrosc., 1993, 25, 1–480 Search PubMed; M. Witanowski and G. A. Webb, Nitrogen NMR, Plenum Press, London, 1973 Search PubMed.
- W. von Philipsborn and R. Müller, Angew. Chem., 1986, 98, 381–482 CrossRef CAS; W. von Philipsborn and R. Müller, Angew. Chem., Int. Ed. Engl., 1986, 25, 383–484 CrossRef.
- G. W. Buchanan, Tetrahedron, 1989, 45, 581–604 CrossRef CAS.
- E. Bojarska-Olejnik, L. Stefaniak, M. Witanowski and G. A. Webb, Bull. Chem. Soc. Jpn., 1986, 59, 3263–3265 CAS.
- H. Günther and P. Schmitt, Naturwissenschaften, 1984, 71, 342–352 CrossRef CAS; M. Schumacher and H. Günther, Chem. Ber., 1983, 116, 2001–2014 Search PubMed.
- H. Wamhoff, in Comprehensive Heterocyclic Chemistry, eds. A. R. Katritzky and C. W. Rees, Pergamon Press, Oxford, 1984, vol. 5, 669–732 Search PubMed; W. Q. Fan and A. R. Katritzky, in Comprehensive Heterocyclic Chemistry II, eds. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon Press, Oxford, 1996, vol. 4, 1–126 Search PubMed.
- H. Quast, T. Hergenröther, K. Banert, E.-M. Peters, K. Peters and H. G. von Schnering, Chem. Ber., 1993, 126, 103–108 CAS.
- K. Banert and S. Groth, Angew. Chem., 1992, 104, 865–867 CAS; K. Banert and S. Groth, Angew. Chem., Int. Ed. Engl., 1992, 31, 866–868 CrossRef.
- K. Banert, Chem. Ber., 1989, 122, 123–128 Search PubMed.
- J. H. Boyer and J. Hamer, J. Am. Chem. Soc., 1955, 77, 951–954 CrossRef CAS.
- P. A. S. Smith and J. H. Boyer, Org. Synth. Coll. Vol., 1963, IV, 75–78 Search PubMed.
- I. Ugi, H. Perlinger and L. Behringer, Chem. Ber., 1958, 91, 2330–2336 Search PubMed.
- E. Leyva, D. Munoz and M. S. Platz, J. Org. Chem., 1989, 54, 5938–5945 CrossRef CAS.
- Y. Ohba, S. Kubo, M. Nakai, A. Nagai and M. Yoshimoto, Bull. Chem. Soc. Jpn., 1986, 59, 2317–2320 CAS.
- D. M. Stout, T. Takaya and A. I. Meyers, J. Org. Chem., 1975, 40, 563–569 CrossRef CAS.
- J. C. Pezzullo and E. R. Boyko, J. Org. Chem., 1973, 38, 168–169 CrossRef CAS.
- W. Kirmse, J. Rode and K. Rode, Chem. Ber., 1986, 119, 3672–3693 Search PubMed.
- K. Banert, Chem. Ber., 1985, 118, 1564–1574 Search PubMed.
- R. L. Eagon, M. A. Ogliaruso and J. P. Springer, J. Org. Chem., 1986, 51, 1544–1547 CrossRef.
- J. R. Johnson and O. Grummitt, Org. Synth. Coll. Vol., 1955, III, 806–807 Search PubMed.
- R. Davis and H. P. Schultz, J. Org. Chem., 1962, 27, 854–857 CAS.
- R. H. Smith, Jr., B. D. Wladkowski, J. E. Taylor, E. J. Thompson, B. Pruski, J. R. Klose, A. W. Andrews and C. J. Michejda, J. Org. Chem., 1993, 58, 2097–2103 CrossRef.
- J. Bourgois, M. Bourgois and F. Texier, Bull. Soc. Chim. Fr., 1978, 485–527 Search PubMed.
- G. Gaudiano, C. Ticozzi, A. Umani-Ronchi and P. Bravo, Gazz. Chim. Ital., 1967, 97, 1411–1422 Search PubMed.
- G. Gaudiano, A. Umani-Ronchi, P. Bravo and M. Acampora, Tetrahedron Lett., 1967, 107–111 CrossRef CAS; H. W. Heine and D. A. Tomalia, J. Am. Chem. Soc., 1962, 84, 993–995 CrossRef CAS.
- R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1966, 99, 475–490 Search PubMed.
- G. D. Buckley, J. Chem. Soc., 1954, 1850–1851 RSC.
- P. Scheiner, Tetrahedron, 1968, 24, 349–356 CrossRef.
- W. Broeckx, N. Overbergh, C. Samyn, G. Smets and G. L'abbé, Tetrahedron, 1971, 27, 3527–3534 CrossRef CAS.
- C. E. Olsen, Acta Chem. Scand., Ser. B, 1974, 28, 425–432 Search PubMed.
- R. Huisgen, L. Möbius and G. Szeimies, Chem. Ber., 1965, 98, 1138–1152 Search PubMed.
- C. S. Rondestvedt, Jr. and P. K. Chang, J. Am. Chem. Soc., 1955, 77, 6532–6540 CrossRef.
- F. Texier and R. Carrié, Bull. Soc. Chim. Fr., 1971, 4119–4128 Search PubMed.
- C. E. Olsen and C. Pedersen, Acta Chem. Scand., 1973, 27, 2271–2278 Search PubMed.
- H. Quast and G. Meichsner, Chem. Ber., 1987, 120, 1049–1058 Search PubMed.
- H. Quast, G. Meichsner and B. Seiferling, Liebigs Ann. Chem., 1986, 1891–1899 Search PubMed.
- K. Hohenlohe-Oehringen, Monatsh. Chem., 1958, 89, 588–596 CAS.
- H. Quast and D. Regnat, Chem. Ber., 1990, 123, 2195–2202 CAS.
- R. F. Bleiholder and H. Shechter, J. Am. Chem. Soc., 1968, 90, 2131–2137 CAS.
- W. R. Peterson, Jr., B. Arkles and S. S. Washburne, J. Organomet. Chem., 1976, 121, 285–291 CrossRef.
- R. Huisgen, L. Möbius, G. Müller, H. Stangl, G. Szeimies and J. M. Vernon, Chem. Ber., 1965, 98, 3992–4013 Search PubMed.
- T. Sasaki, S. Eguchi, M. Yamaguchi and T. Esaki, J. Org. Chem., 1981, 46, 1800–1804 CrossRef CAS.
- R. E. Banks and A. Prakash, J. Chem. Soc., Perkin Trans. 1, 1974, 1365–1371 RSC.
- P. Scheiner, J. Org. Chem., 1965, 30, 7–10 CAS.
- P. Scheiner, Tetrahedron, 1968, 24, 2757–2766 CrossRef CAS.
- P. Scheiner, J. H. Schomaker, S. Deming, W. J. Libbey and G. P. Nowack, J. Am. Chem. Soc., 1965, 87, 306–311 CrossRef CAS.
- K. B. Becker and M. K. Hohermuth, Helv. Chim. Acta, 1979, 62, 2025–2036 CrossRef CAS.
- R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1967, 100, 2494–2507 Search PubMed.
- G. Bianchetti, D. Pocar and P. D. Croce, Rend. Ist. Lomb. Sci. Lett., 1965, A99, 316–335 (Chem. Abstr., 1966, 65, 3860h) Search PubMed.
- A. Mustafa, S. M. A. D. Zayed and S. Khattab, J. Am. Chem. Soc., 1956, 78, 145–149 CrossRef CAS.
- H. Möhrle and H. Dwuletzki, Chem. Ber., 1986, 119, 3591–3599 Search PubMed.
- P. Scheiner, J. Org. Chem., 1967, 32, 2022–2023 CrossRef CAS.
- A. L. Logothetis, J. Am. Chem. Soc., 1965, 87, 749–754 CrossRef CAS.
- P. S. Engel, L. Pan, K. H. Whitmire, I. Guzman-Jimenez, M. R. Willcott and W. B. Smith, J. Org. Chem., 2000, 65, 1016–1021 CrossRef CAS.
- M. Witanowski, L. Stefaniak and G. A. Webb, Annu. Rep. NMR Spectrosc., 1977, 7, 209 Search PubMed; M. Witanowski, L. Stefaniak and G. A. Webb, Annu. Rep. NMR Spectrosc., 1981, 11B, 103 Search PubMed; T. B. Patrick and R. P. Willaredt, J. Org. Chem., 1983, 48, 4415–4416 CrossRef CAS; A. Lyčka and P. Vetešník, Collect. Czech. Chem. Commun., 1984, 49, 963–969 CAS; D. E. V. Wilman, Magn. Reson. Chem., 1990, 28, 729–731 CAS; J. V. Jollimore, K. Vaughan and D. L. Hooper, J. Org. Chem., 1996, 61, 210–214 CrossRef CAS; R. E. Brown and G. D. Mendenhall, J. Phys. Chem. A, 1998, 102, 8537–8540 CrossRef CAS.
- B. D. Wladkowski, R. H. Smith and C. J. Michejda, J. Am. Chem. Soc., 1991, 113, 7893–7897 CrossRef CAS.
- R. O. Duthaler and J. D. Roberts, J. Am. Chem. Soc., 1978, 100, 3889–3895 CrossRef CAS.
- S. Berger, S. Braun and H.-O. Kalinowski, NMR-Spektroskopie von Nichtmetallen, Band 2, 15N-NMR-Spektroskopie, Georg Thieme, Stuttgart, 1992, pp. 4, 11 and 17 Search PubMed.
- L. P. Hammett, Physical Organic Chemistry, 2nd edn., McGraw-Hill, New York, 1970 Search PubMed.
- R. W. Taft, Jr., J. Phys. Chem., 1960, 64, 1805–1815 CrossRef.
| This journal is © The Royal Society of Chemistry 2002 |