Photooxidation of n-heptanal in air: Norrish type I and II processes and quantum yield total pressure dependency
Jovan M.
Tadić
*a,
Ivan O.
Juranić
b and
Geert K.
Moortgat
c
aCenter for Chemistry IChTM, POB 815, Studentski trg 12-16, 11001 Belgrade, Yugoslavia. E-mail: jtadic@Eunet.yu; Fax: ++381-11-639-827; Tel: ++381-11-3236-603
bFaculty of Chemistry, University of Belgrade, POB 158, 11001 Belgrade, Yugoslavia. Fax: ++381-11-639-827; Tel: ++381-11-637-405
cMax-Planck-Institut für Chemie, Atmospheric Chemistry Department, Postfach 3060, 55020 Mainz, Germany. E-mail: moo@mpch-mainz.mpg.de; Fax: ++49-6131-305-436; Tel: ++49-6131-305-476
First published on 7th December 2001
Abstract
Dilute mixtures of n-heptanal in synthetic air (up to 100 ppm) were photolyzed with fluorescent UV lamps (275–380 nm) at 298 K. The main photooxidation products, identified and quantitatively analyzed by FTIR spectroscopy, were pent-1-ene, CO, vinyl alcohol and ethanal. The photolysis rates and the absolute quantum yields Φ were found to be slightly dependent on the total pressure. At 100 Torr, Φ100
= 0.36 ± 0.03, whereas at 700 Torr the total quantum yield was Φ700
= 0.31 ± 0.01. The results may be explained by the collisional deactivation of photoexcited molecules. Two decomposition channels were identified: the radical channel C6H13CHO → C6H13
+ HCO, and the molecular channel C6H13CHO → C5H10
+ CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHOH, having the absolute quantum yields of 0.031
and 0.118 at 700 Torr. The product CH2CHOH tautomerizes to ethanal.
CHOH, having the absolute quantum yields of 0.031
and 0.118 at 700 Torr. The product CH2CHOH tautomerizes to ethanal.
Introduction
The role of aldehydes in the formation of photochemical smog, peroxyacetyl nitrate (PAN), and regional (tropospheric) ozone is well known. Photodissociation of aldehydes represents an important source of free radicals in the lower atmosphere, and thus may significantly influence the atmospheric oxidation capacity.1 Aldehydes are widely used in industry and are the products of an incomplete combustion of petroleum fuels. Smaller alkyl aldehydes are also products of the atmospheric photooxidation of hydrocarbons, ethers, alcohols, and other organic compounds. Vegetation, biomass and other living organisms emit many of these compounds: n-hexanal and higher aldehydes have been observed in ambient air and in emissions of various plants, especially grasses, with comparable emission rates to the monoterpenes.2 The examination of longer chain aldehydes has recently come into the research focus, providing quantum yield and mechanistic data necessary for atmospheric modeling.3–6 This is the first reported study on the photolysis of n-heptanal.Aldehydes absorb in the near UV range, and dissociate upon absorption of light. Aliphatic aldehydes exhibit a weak absorption band in the wavelength range 240–360 nm as a result of a symmetry forbidden n–π* transition.7,8 There have been a number of studies devoted to the photodissociation of the simplest alkyl aldehydes, such as HCHO, CH3CHO, C2H5CHO,9–17 and a recently growing number devoted to longer chain aldehydes, such as C3H7CHO, C4H9CHO,3,18 isopentanal (3-methylbutanal) and tert-pentanal (2,2-dimethylpropanal)5 and n-hexanal.6
The n-hexanal photodissociation pattern6 and reaction schemes for n-pentanal3 suggest that processes (1) and (2) play an important role in the photolysis of longer chain aldehydes:
| n-heptanal + hν → n-C6H13 + HCO (Norrish Type I) | (1) |
|
→ C5H10
+
[CH2CHOH] (Norrish Type II)
| (2) |
Process (1) represents the fragmentation into free radicals, with an enthalpy of around 350 kJ mol−1 corresponding to a photochemical threshold of around 340 nm.3 Process (2), which is common to molecules with a γ-hydrogen atom, is an intramolecular rearrangement with enthalpy around 80 kJ mol−1 (λ ≤ 1454 nm).3 The enthalpy change for process (2) was calculated assuming that the keto-form of acetaldehyde is formed in the primary step. This assumption is not correct, and, therefore, the enthalpy change for this reaction should be adjusted for the difference between the heats of formation of the enol and keto forms of acetaldehyde (the pK value for the keto–enol equilibrium is ∼7 and the corresponding ΔG value is ∼18 kJ mol−1).19 The total contribution of the Norrish I and II processes in the case of n-butanal, n-pentanal and n-hexanal photolysis is, however, less than 100%, indicating a further reaction pathway (the calculation is made on carbon balance data).4,6
In this paper, the results obtained from the photolysis of small quantities of n-heptanal in air, using wide band emission lamps, are reported. We have investigated products and absolute quantum yields in the pressure range 100 to 700 Torr.
The photooxidation experiments were carried out in a long-path quartz cell with detection of precursors and products by FTIR spectroscopy. After identification and quantification of the products, a mechanistic description of the photooxidation/photolysis is proposed. From the measured decay rate of the starting material, and from knowledge of the absorption spectrum, overall quantum yields for the photolysis were calculated for various pressures.
Results and discussion
Observed products and mechanism
Fig. 1 shows the FTIR spectra of a mixture of 100 ppm n-heptanal in synthetic air, before and after the photolysis. Major products observed are CO (νmax = 2037–2235 cm−1), pent-1-ene (913.7, 1000, 1461, 1646, 2810–3041, 3087.8 cm−1), ethenol (vinyl alcohol), which is the enol form of ethanal (947.6, 1078, 1118.3, 1259.8 cm−1) and ethanal (1348.5–1355.5 cm−1).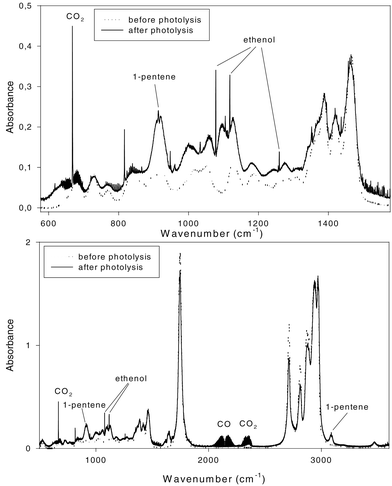 | ||
| Fig. 1 FTIR spectrum of 100 mTorr n-heptanal, before and after photolysis (6 TL/12 lamps, 100 Torr synthetic air). Major products, CO, pent-1-ene, and ethenol, and CO2 which is formed as a by-product, are marked (exact positions of the peaks are given in the main text). | ||
Fig. 2 shows the concentration–time profiles for two products used for the estimation of Norrish type I/II absolute yields and ratio. The only peak of pent-1-ene that does not interfere with other presented compounds, and which was used for the quantification, is fairly weak, resulting in a large uncertainty in the pent-1-ene concentration at the beginning of the photolysis and so the first two points have been omitted from the plots.
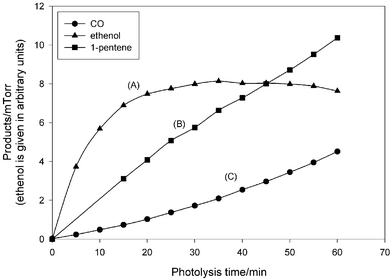 | ||
| Fig. 2 Photolysis of n-heptanal: time profile-variation of the partial pressures of the products. The curve of CO (C) shows that CO is not only the primary product, but also a product of the reactions following the primary step. Ethenol partial pressure reaches a maximum and then starts to decrease by conversion to ethanal. | ||
Pent-1-ene is formed exclusively as a primary product in the reaction. Ethanal is a secondary product arising from ethenol conversion, and undergoes further photolysis, which was described in our previous work on n-pentanal.4 This time only the identification of ethanal was done.
Reaction (1) gives two radicals, which immediately react with oxygen. One of the products, formyl radical HCO is quantitatively converted to CO and HO2,9,20,21 according to reaction (3).
| HCO + O2 → HO2 + CO | (3) |
This process is followed through the rate of production of CO (2094–2096 cm−1). The n-hexyl radicals are oxidized forming n-hexylperoxyl radicals [reaction (4)].
| n-C6H13 + O2 + M → n-C6H13O2 + M | (4) |
The n-C6H13O2 radicals then react with HO2, recombine or disproportionate, to produce a variety of products, reactions (5)–(8).22,23
| n-C6H13O2 + HO2 → n-C6H13OOH + O2 | (5) |
| 2 n-C6H13O2 → 2 n-C6H13O + O2 | (6) |
| →n-C6H13OH + n-C5H11CHO + O2 | (7) |
| n-C6H13O + O2 → n-C5H11CHO + HO2 | (8) |
However due to the estimated low reaction rate constant for the recombination and disproportionation, reactions (6) and (7),23 the major product expected is n-hexyl hydroperoxide (n-C6H13OOH). This peroxide was not observed in the FTIR spectrum. The concentrations expected either were under the detection limit (∼1 mTorr) or the signals interfered with other compounds. Also no evidence of n-hexanal produced viachannels (7) and (8) was observed.
Pent-1-ene is a stable molecular species and was used to calculate the rate of the Norrish type II process (2). The co-product of the reaction (2) is ethenol CH2CHOH, which tautomerizes slowly to ethanal. This is clearly seen in Fig. 2, where the peak integral–time profile of the enol product displays the behavior of a primary product involved in a secondary reaction. Ethenol concentration reaches a maximum after ∼35 minutes photolysis, and its subsequent decay is caused by its conversion to ethanal. Fig. 3 shows profiles of product concentrations (partial pressures) of CO and pent-1-ene versus loss of n-heptanal. According to the suggested mechanism, the yield of pent-1-ene should be identical to the sum of the yields of ethenol and ethanal, since all compounds are products of the same decomposition channel. CO coming
from the reaction of HCO radical with oxygen can be treated as a primary product, but increases of the yield at long conversion times are observed, which can be attributed to the photolysis of secondary products such as ethanal, n-hexanal, etc. Theoretically, another possible source of CO could be reaction (9).
| n-C7H14O + hν → n-C6H14 + CO | (9) |
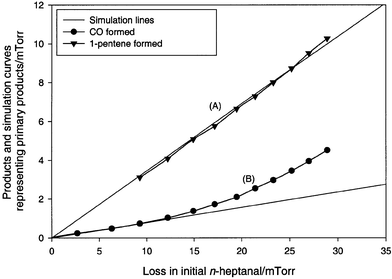 | ||
| Fig. 3 Photolysis of n-heptanal—products formed versus loss of n-heptanal. See comments on Fig. 2. | ||
The stable product of this reaction, n-hexane, was not identified in our experiments. The relative probability for the n-heptanal molecule to undergo decomposition by two detected channels is shown in Table 1. The probability is deduced from the ratio of primary formed CO (Norrish Type I), and pent-1-ene (Norrish Type II).
| Total pressure/Torr | Norrish type I | Norrish type II |
|---|---|---|
| 100 | 24.89 | 75.11 |
| 100 | 23.87 | 76.13 |
| 100 | 27.79 | 72.21 |
| 100 | 22.00 | 78.00 |
| 100 | 17.00 | 83.00 |
| 300 | 14.85 | 85.15 |
| 300 | 15.16 | 84.96 |
| 300 | 14.75 | 85.25 |
| 500 | 15.24 | 84.76 |
| 500 | 19.96 | 80.04 |
| 500 | 17.29 | 82.71 |
| 700 | 21.46 | 78.54 |
| 700 | 19.11 | 80.89 |
| 700 | 20.10 | 79.90 |
| Average | 21.0 ± 6.3 | 79.0 ± 6.3 |
Assuming that the formation of CO2 is an artifact, the sum of both processes, Norrish type I and II, is 48 ± 7% (Table 2), indicating the presence of other unidentified product channels.
| Total pressure/Torr | Norrish type I (Δprimary CO/Δheptanal) | Norrish type II (Δpent-1-ene/Δheptanal) |
|---|---|---|
| 100 | 11.95 | 36.05 |
| 100 | 11.46 | 36.54 |
| 100 | 13.34 | 34.66 |
| 100 | 10.56 | 37.44 |
| 100 | 8.16 | 39.84 |
| 300 | 7.13 | 40.87 |
| 300 | 7.28 | 40.78 |
| 300 | 7.08 | 40.92 |
| 500 | 7.32 | 40.68 |
| 500 | 9.58 | 38.42 |
| 500 | 8.30 | 39.70 |
| 700 | 10.30 | 37.70 |
| 700 | 9.17 | 38.83 |
| 700 | 9.65 | 38.35 |
| Average | 10.08 ± 8.00 | 37.92 ± 2.80 |
n-Heptanal molecule could undergo photocyclisation, forming cis/trans isomers of 2-propylcyclobutanol (Scheme 1). This mechanism is observed in the photolysis of n-pentanal,24 and some ketones.7 This compound was not detected in our experiments.
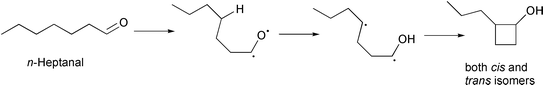 | ||
| Scheme 1 | ||
Absolute quantum yields
One of the objectives of the study was to determine the dependence of the absolute quantum yield on the total pressure. n-Butanal was used as the actinometer, with absolute quantum yields recently reported.4 From the decay of n-heptanal concentration (partial pressure), the photolytic rate constants were deduced for the different pressures by plotting the natural logarithm of concentration versus time (first order decay), and performing a least-squares fit. From these results, overall quantum yields were calculated according to eqn. (1) using the overlap integrals of n-heptanal and n-butanal spectra (integral equals unity) within the range of the TL/12 emission (275–380 nm, see Fig. 4). In all cases an absolute quantum yield dependency on the total pressure was observed. Measurements of all n-alkanal cross sections from C2–C9 have been performed very recently by Zabel.25 This study reveals that the UV spectra of the aldehydes from n-butanal upwards are practically identical.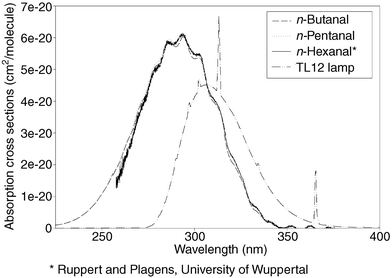 | ||
| Fig. 4 Emission spectrum of photolysis lamps (TL12/Philips) and cross section of n-hexanal,24n-pentanal and n-butanal.4 The figure explains why TL12 lamps were chosen for photolysis, since there is a good overlap of emission and absorption spectrum. | ||
The absolute quantum yield data are summarized in Table 3 for all experiments performed at total pressures of 100, 300, 500 and 700 Torr and are also shown in the form of a Stern–Volmer plot in Fig. 5. Since the intercept at zero pressure is not equal to 1 (the intercept is 2.4 ± 0.1), which should be the case if collisional deactivation was the only relaxation process besides the photodecomposition, it seems very probable that there are other energy-dissipating processes (the triplet state of n-heptanal could deactivate by phosphorescence, etc.).6 The interaction of the photoexcited molecules with the walls resulting in the relaxation to the ground state is of minor importance because of the large volume to surface ratio of the reaction cell. The slope of the fit, described by 1.169 × 10−3 × P, corresponds to the sensitivity of absolute quantum yield to the total pressure P (in torr).
| Total pressure/Torr | Absolute quantum yield | |||
|---|---|---|---|---|
| n-Heptanal | n-Hexanal6 | n-Pentanal4 | n-Butanal4 | |
| 100 | 0.39 | – | – | – |
| 100 | 0.35 | – | – | – |
| 100 | 0.33 | – | – | – |
| 100 | 0.41 | – | – | – |
| 100 | 0.45 | – | – | 0.47 |
| 100 | 0.37 | 0.43 | 0.40 | 0.50 |
| 100 | 0.42 | 0.41 | 0.44 | 0.47 |
| 100 | 0.40 | 0.44 | 0.36 | 0.47 |
| 0.39 ± 0.06 | 0.43 ± 0.02 | 0.40 ± 0.04 | 0.48 ± 0.02 | |
| 300 | 0.38 | – | – | – |
| 300 | 0.36 | – | – | – |
| 300 | 0.36 | – | – | – |
| 300 | 0.41 | 0.41 | 0.37 | 0.43 |
| 300 | 0.41 | 0.42 | 0.39 | 0.44 |
| 300 | 0.36 | 0.43 | 0.36 | 0.44 |
| 0.38 ± 0.03 | 0.42 ± 0.01 | 0.38 ± 0.02 | 0.44 ± 0.01 | |
| 500 | 0.32 | – | – | – |
| 500 | 0.32 | – | – | – |
| 500 | 0.32 | – | – | – |
| 500 | 0.37 | 0.43 | 0.36 | 0.30 |
| 500 | 0.33 | 0.38 | 0.36 | 0.37 |
| 500 | 0.37 | 0.41 | 0.33 | 0.37 |
| 0.32 ± 0.05 | 0.40 ± 0.03 | 0.35 ± 0.02 | 0.35 ± 0.05 | |
| 700 | 0.31 | – | – | – |
| 700 | 0.30 | – | – | – |
| 700 | 0.31 | – | – | – |
| 700 | 0.31 | 0.40 | 0.35 | 0.32 |
| 700 | 0.29 | 0.37 | 0.34 | 0.33 |
| 700 | 0.31 | 0.37 | 0.33 | 0.32 |
| 0.31 ± 0.02 | 0.38 ± 0.02 | 0.34 ± 0.01 | 0.32 ± 0.01 | |
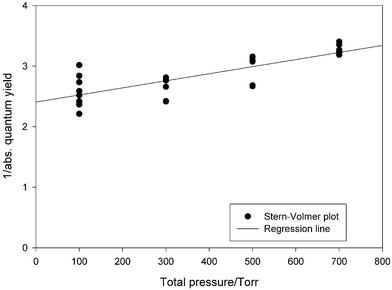 | ||
| Fig. 5 The pressure dependency of 1/(absolute quantum yields) in n-heptanal photolysis, at different total pressures of synthetic air (Stern–Volmer plot). If collisional deactivation is the only relaxation process, one should expect that the intercept on the ordinate should be unity. This is not the case, indicating that one or more other energy dissipating processes which form photoexcited molecules are taking place. | ||
The total quantum yield Φtot can be calculated from the following equation 1/Φtot = 2.408 + (1.169 × 10−3 × P). Using the estimated relative quantum yield from Tables 1 and 2, it is possible to calculate the absolute contribution at atmospheric conditions (700 Torr): for Norrish type I (radical) process, ϕ(I) = 0.031 ± 0.001 and for Norrish type II (molecular) ϕ(II) = 0.118 ± 0.001 (total quantum yield being 0.31, and the contribution of both decomposition channels being 48 ± 7%).
Comparison with other aldehydes (Table 3) shows that n-pentanal, n-hexanal and n-heptanal have similar dependencies on the total pressure of synthetic air.4,6 The sensitivity of the quantum yield to the total pressure (Torr) is described by the slope of the curve in the Stern–Volmer plot, and was 1.169 × 10−3, 4.75 × 10−4, 7.77 × 10−4 and 1.93 × 10−3, for n-heptanal, n-hexanal, n-pentanal and n-butanal, respectively.4,6n-Heptanal is more or less in line with n-hexanal and n-pentanal; n-butanal sensitivity is somewhat higher which is also consistent with the substantially different Norrish type I/II ratio. This may point to different spin states distribution upon photoexcitation.
Future work on the photolysis of the longer chain aldehydes should concentrate on the determination of the spin states of photoexcited molecules as precursors of Norrish type I and II processes, and on the analysis of other possible products, since ∼50% of the initial aldehyde cannot be accounted for by Norrish type I and II reaction mechanisms. There is no conclusive evidence for the formation of cyclobutanol derivatives and further examinations on the subject should be done. Possible examinations of the absolute quantum yield dependency on the low pressures of pure oxygen may reveal new information. The triplet ground electronic state of oxygen means that reaction pathways proceeding from the lowest triplet of the aldehyde (T1) would probably be quenched more efficiently than the ones proceeding from the vibrationally excited ground singlet (S0*), or eventually from the first excited singlet (S1). Thus a stronger dependence of the decomposition parameters on oxygen pressure may be observed.
Atmospheric implications
The main degradation processes of carbonyl compounds are controlled by photolysis and by the reaction with OH radicals. The atmospheric lifetime of n-heptanal can be estimated from the knowledge of the OH reaction rate constant and the photodissociation rate. Unfortunately the rate constant for OH reaction is not available. The rate constants for the OH reactions of the homologous aldehydes, n-butanal,26,27n-pentanal,24 and n-hexanal were estimated to be 2.35 × 10−11, 2.7 × 10−11 and 1.9 × 10−11 cm3 molecule−1 s−1, respectively. Although no data on the rate constant of the n-heptanal OH reaction are available, it is expected to be in line with these measurements, resulting in a reactive lifetime of 5–7 hours under atmospheric conditions (average noon-time OH concentration of 2 × 106 molecules cm−3 was used).Maximum daytime photolysis rates for n-butanal and n-pentanal were estimated during in-situ measurements in the photochemical outdoor reactor,24 as kph,b = (1.0 ± 0.2) × 10−5 s−1 for n-butanal and kph,p = (1.6 ± 0.15) × 10−5 s−1 for n-pentanal, corresponding to a photolytic lifetime of 28 hours and 17 hours, respectively. Our previous work on n-pentanal4 and this study reveal similar behavior of n-heptanal, n-hexanal and n-pentanal photolysis parameters under laboratory conditions, including absolute quantum yield values, sensitivity to total pressure, and Norrish type I/II ratio. This would indicate that the dominant removal process in the lower troposphere for n-heptanal, like for other homologous aldehydes, is the reaction with OH radicals, but that photolytic processes still take some part in the degradation.
The absolute radical yield under atmospheric conditions can be estimated for n-heptanal as 10.08 × 0.31 = 3.1%. If quantum yield values for aldehydic compounds are not known (e.g. for use in atmospheric modeling), it is common to assume that they are unity, which is seldom the case as shown in this, and previous studies.3–6,24 This often leads to an overestimation of the calculated radicals produced by photolysis processes.
Conclusions
In this work we achieved several goals.We identified the primary products of n-heptanal photolysis following Norrish type I and II decomposition channels, detectable in the IR region under the experimental conditions.
We quantified the products and partially deduced the primary photodecomposition pattern.
We determined the absolute quantum yields at different pressures, providing values necessary for atmospheric modeling, at the same time examining the influence of the total pressure on the absolute quantum yield and evaluating the importance of collisional deactivation, and indicating other relaxation channels.
There is evidence that for up to C4 aldehydes the decomposition upon absorption of light mainly follows the free radical channel (Norrish type I), forming a formyl radical and alkyl radicals.4,6,24 Higher than C4 aldehydes, starting with n-pentanal, mainly decompose by internal rearrangement of the molecule (Norrish type II), forming vinyl alcohol, and the corresponding alk-1-ene. Our results on n-heptanal photolysis and previous work support this pattern. However, the decrease of the absolute yields of Norrish type I and II processes indicates that other, so far unidentified, processes become more and more important in the photolysis of longer chain aldehydes.
Experimental
The apparatus employed in this work has been described elsewhere28,29 and so will only be briefly discussed here. The central part of the apparatus is a 44.2 liter (1.40 m length and 20 cm diameter) quartz cell equipped with two independent sets of White-optic mirror arrangements. Sapphire-coated aluminium mirrors were used in the infrared region (l = 33.6 m) for the measurements of the educts and products. Infrared spectra at 0.5 cm−1 resolution (450–4000 cm−1) were measured with a Bomem DA8-FTIR spectrometer. For the UV measurements the same diode array detector as previously described was used.28 This method provides the possibility of simultaneous detection and monitoring of all the IR-active products and the starting material.Photolysis was achieved with six radially mounted lamps, TL/12-sunlamps (Philips 40 W TL/12 lamps λ = 275–380 nm). Spectra were taken every 5 min with a total irradiation time of 60 min. The reaction was followed until the aldehyde concentration had decreased by 30%.
The use of a continuous broad band light source allows only the determination of an integral, effective quantum yield Φint for the photoactive spectral region. Quantum yields were calculated according to the following equation (for carbonyl compound, C, and actinometer, Act):
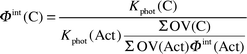
For all TL/12-experiments, n-butanal was used as the actinometer (with ϕ = 0.48 ± 0.02, 0.44 ± 0.01, 0.35 ± 0.05, and 0.32 ± 0.01 at 100, 300, 500, and 700 Torr, respectively). The quantum yield is the only unknown parameter in the equation. The photolysis rate for the compound Kphot(C) could be directly measured, and the terms ΣOV(C) and ΣOV(Act) represent the calculated overlap of lamp emission and absorption spectrum of substrate. Fig. 4 displays the emission spectra of the lamps and the spectra of homologous aliphatic aldehydes.
The knowledge of the UV absorption spectra of n-heptanal was a basic prerequisite for these experiments. The absorption spectrum has been recently measured by Zabel,25 and is practically identical to the spectra of homologous aldehydes, n-butanal and n-pentanal.4,24,30 The spectrum displays a broad absorption band between 250 and 350 nm, with a maximum absorption observed at 295 nm with a cross section of 6.0 × 10−20 cm2 molecule−1.
Experiments were carried out at room temperature (298 K), at pressures between 100 and 700 Torr (1 Torr = {101325/760}Pa), with an initial aldehyde concentration of approx. 100 ppm. Qualitative and quantitative data evaluation was carried out by comparing the product spectra with reference spectra obtained in the same cell and using calibration curves at corresponding pressures and resolution.
Carbonyl compounds were obtained from Sigma-Aldrich Company with purity higher than 95%. Before use, all samples were degassed by several freeze–pump–thaw cycles. The purity of the compounds was checked by FTIR spectral measurements and no impurities were found. The conversion of vinyl-alcohol and ethanal is 1 ∶ 1 (keto–enol tautomerism) according to reaction (10).
|
CH2CH–OH ⇌ CH3–CHO
| (10) |
References
- T. E. Graedel, L. A. Farrow and T. A. Weber, Atmos. Environ., 1976, 10, 1095 CrossRef CAS; D. Grosjean, Environ. Sci. Technol., 1982, 16, 254 CAS; B. J. Finlayson-Pitts and J. N. Pitts, Atmospheric Chemistry, John Wiley, New York, 1986 Search PubMed.
- S. Owen, R. Boissard, R. A. Street, S. C. Duckam, O. Csiky and C. N. Hewitt, Atmos. Environ., 1997, 31, 101 CrossRef CAS; W. Kirstine and I. Galbally, J. Geophys. Res., 1998, 103, 10603 CrossRef.
- J. T. Cronin and L. Zhu, J. Phys. Chem. A, 1998, 102, 10274 CrossRef.
- J. Tadic, I. Juranic and G. K. Moortgat, J. Photochem. Photobiol. A: Chem., 2001, 143, 169 Search PubMed.
- L. Zhu, J. T. Cronin and A. Narang, J. Phys. Chem. A, 1999, 103, 7248 CrossRef CAS.
- J. Tadic, I. Juranic and G. K. Moortgat, Molecules, 2001, 6, 287 Search PubMed.
- J. G. Calvert and J. N. Pitts, Photochemistry, John Wiley, New York, 1966, pp. 372–375 Search PubMed.
- E. K. C. Lee and R. S. Lewis, Adv. Photochem., 1980, 12, 1 Search PubMed.
- G. K. Moortgat, W. Seiler and P. Warneck, J. Chem. Phys., 1983, 78, 1185 CrossRef CAS.
- Y. Carmely and A. Horowitz, Int. J. Chem. Kinet., 1984, 16, 1585 CrossRef CAS.
- C. B. Moore and J. C. Weishaar, Annu. Rev. Phys. Chem., 1983, 34, 525 CrossRef CAS.
- P. Ho, D. J. Bamford, R. J. Buss, Y. T. Lee and C. B. Moore, J. Chem. Phys., 1982, 76, 3630 CrossRef CAS.
- A. Horowitz and J. G. Calvert, J. Phys. Chem., 1982, 86, 3105 CrossRef CAS.
- H. Meyrahn, G. K. Moortgat and P. Warneck, Presented at the 15th Informal Conference on Photochemistry, Stanford, CA, July 1982.
- P. B. Shepson and J. Heicklen, J. Photochem., 1982, 19, 215 Search PubMed.
- J. Heicklen, J. Desai, A. Bahta, C. Harper and R. Simonaitis, J. Photochem., 1986, 34, 117 Search PubMed.
- A. C. Terentis, P. T. Knepp and S. H. Kable, J. Phys. Chem., 1995, 99, 12704 CrossRef CAS.
- S. Forgeter, T. Berces and S. Dobe, Int. J. Chem. Kinet., 1979, 11, 219 CrossRef CAS.
- R. P. Bell and P. W. Smith, J. Chem. Soc. (B), 1966, 241 RSC.
- A. Horowitz and J. G. Calvert, Int. J. Chem. Kinet., 1978, 10, 805 CrossRef CAS.
- A. Horowitz, F. Su and J. G. Calvert, Int. J. Chem. Kinet., 1978, 10, 1099 CrossRef CAS.
- R. Atkinson, J. Phys. Chem. Ref. Data, 1994 , Monograph 2.
- P. D. Lightfoot, R. A. Cox, J. N. Crowley, M. Destriau, G. D. Hayman, M. E. Jenkin, G. K. Moortgat and F. Zabel, Atmos. Environ., 1992, 26A, 1805 CAS.
- G. K. Moortgat, Final report on EU project RADICAL: “Evaluation of radical sources in atmospheric chemistry through chamber and laboratory studies”ENV4-CT97-0419, March 2000 Search PubMed.
- F. Zabel, Reaktionswege von Alkoxyradikalen unter atmosphärischen Bedingungen und UV-Absorptionsspektren von Carbonylverbindungen, in K.H. Becker (coordinator), Annual report 1998 on BMBF-project TFS/LT3, FKZ 07TFS30, 1999, p. 69.
- D. H. Semmes, H. R. Ravishankara, C. A. Gump-Perkins and P. H. Wine, Int. J. Chem. Kinet., 1985, 17, 303 CrossRef CAS.
- J. A. Kerr and D. W. Sheppard, Environ. Sci. Technol., 1981, 15, 960 CAS.
- G. K. Moortgat, R. A. Cox, G. Schuster, J. P. Burrows and G. S. Tyndall, J. Chem. Soc., Faraday Trans. 2, 1989, 85, 809 RSC.
- W. H. Raber and G. K. Moortgat, Adv. Ser. Phys. Chem., 3, “Progress and Problems in Atmospheric Chemistry”, Ed. J. R. Barker, Word Scientific Publishing Co., Singapore, 1995, pp. 318–373 Search PubMed.
- R. D. Martinez, A. A. Buitrago, N. W. Howell, C. H. Hearn and J. A. Joens, Atmos. Environ., 1992, 26, 785.
| This journal is © The Royal Society of Chemistry 2002 |