Products from the reaction of C60F18 with sarcosine and aldehydes: the Prato reaction
Xian-wen
Wei
a,
Anthony G.
Avent
a,
Olga V.
Boltalina
b,
Joan M.
Street
c and
Roger
Taylor
*a
aThe Chemistry Laboratory, CPES School, Sussex University, Brighton, UK BN1 9QJ
bChemistry Department, Moscow State University, Moscow 119899, Russia
cChemistry Department, The University, Southampton, UK SO17 1BJ
First published on 21st November 2001
Abstract
Various pyrrolidinofullerenes (both mono- and bis-addition products) have been isolated from the reaction between toluene solutions of C60F18, N-methyl-2-aminoethanoic acid (sarcosine) and either formaldehyde or benzaldehyde. Both mono- and bis-addition products were obtained and either partially of fully characterised. The main mono-addition product from the reaction with formaldehyde was also obtained from reactions with the other aldehydes, indicating that formaldehyde may be present in sarcosine, or generated from it through in situ oxidation by the fullerene. The main mono-addition product from reaction of C60F18 with toluene solutions of sarcosine and benzaldehyde was obtained also in the absence of benzaldehyde. This anomaly was traced to the presence of benzaldehyde (0.05%) in HPLC grade toluene, which may be increased by oxidation of toluene during the reaction conditions.
As part of a programme to investigate the utility of C60F18 for the preparation of donor–acceptor diads, we are carrying out preliminary studies of archetypal reactions that have been employed for this purpose with [60]fullerene. The [3 + 2] dipolar cycloaddition reaction between aldehydes, amino acids and [60]fullerene (Prato reaction)1–13 has been used very extensively in particular in this connection. We have therefore embarked on a programme of investigating such reactions with C60F18 (a better electron acceptor than [60]fullerene). In the first instance we have used formaldehyde and benzaldehyde as the aldehydes in the reaction with N-methyl-2-aminoethanoic acid (sarcosine), and have isolated a number of mono- and bis-addition products. These cycloadditions also take place in the absence of added aldehyde due possibly to the presence of formaldehyde in the sarcosine, or generated from the latter by fullerene-catalysed oxidation. Similar anomalous cycloaddition takes place (to give the main product formed normally in the presence of benzaldehyde) just on heating sarcosine, toluene and [60]fullerene.
Experimental
N-Methyl-2-aminoethanoic acid (1 mg) and paraformaldehyde (1 mg), added to a solution of C60F18 (5 mg in 20 cm3 of HPLC grade toluene) under argon, were heated under reflux for 2 h. The crude product was filtered and separated by HPLC (high pressure liquid chromatography) using a 10 × 250 mm Cosmosil Buckyprep column, eluted with toluene at a flow rate of 4.7 ml min−1. Fractions were collected with retention times of 43, 29, 27, 21, 18.4, 12.9, 12.4, 10.9 and 10.2 min. The EI mass spectra of all of these fractions showed only the presence of C60F18 due to the retro reaction.A second reaction was carried out using benzaldehyde instead of paraformaldehyde. Products were separated having retention times of 44.6, 38, 26.5, 22.0, 18.8, 16.0, 13.1, 11.7, 8.7, 6.9, and 6.5 min; the 38 min fraction was recovered C60F18, and the 22 min fraction proved to be identical to the 21 min fraction obtained with formaldehyde.
Further reactions were carried out as above but using: (i) C60F18, sarcosine and toluene; (ii) C60, sarcosine and benzene; (iii) C60, sarcosine, benzaldehyde and toluene; (iv) C60, sarcosine, and toluene.
1H NMR spectra recorded at 500 MHz, and 19F NMR at 376.5 MHz.
Results and discussion
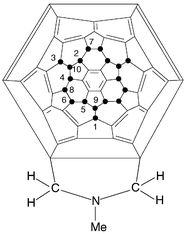
Before interpreting the individual results we consider the various bonds across which addition can occur. These are a–d on the Schlegel diagram (Fig. 1). Thus there are four mono-addition sites, and for reaction with sarcosine and formaldehyde, two products will be symmetrical (a and c addition), and two will be unsymmetrical (b and d addition). It is reasonable to assume, that reaction across bond d will be sterically unfavourable. | ||
| Fig. 1 Schlegel diagram for C60F18 (● = F), showing the four different bonds (a–d) across which cycloaddition can occur; equivalent bonds are a1 etc. | ||
Bis-addition of these reagents to [60]fullerene occurs readily and has been studied in detail.14 For bis-addition to C60F18, symmetrical products can arise from addition across bonds (a + a1), (a + c1), (b + b1), (b + b3), (b + b5), (c + c1), (d
+
d1), (d
+
d3), and (d
+
d5), whilst the unsymmetrical products arise from addition across bonds (a + b), (a + b1), (a + b2), (a + c), (a
+
d), (a
+
d1), (a
+
d2), (b + b2) [![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) (b + b4)], (b + c), (b + c1), (b + c2), (b
+
d), (b
+
d1), (b
+
d2), (b
+
d3), (b
+
d4), (b
+
d5), (c
+
d1), (c
+
d2), (c
+
d3), (d
+
d2), (d
+
d4). Since those involving d additions (italicised) can be disregarded for steric reasons, we can expect in principle up to six symmetrical and eight unsymmetrical bis-addition products.
(b + b4)], (b + c), (b + c1), (b + c2), (b
+
d), (b
+
d1), (b
+
d2), (b
+
d3), (b
+
d4), (b
+
d5), (c
+
d1), (c
+
d2), (c
+
d3), (d
+
d2), (d
+
d4). Since those involving d additions (italicised) can be disregarded for steric reasons, we can expect in principle up to six symmetrical and eight unsymmetrical bis-addition products.
The reaction involving formaldehyde
The products obtained and their characterisation (where possible) are as follows. | ||
| Fig. 2 1H NMR spectrum for 1. | ||
The 19F NMR spectrum (Fig. 3) consists of ten lines at δF −130.62 (1 F, br s), −134.38 (2 F, br s), −134.50, (2 F, br s), −135.65 (2 F, br s), −135.98 (2 F, br s), −146.05 (2 F, d, J 30 Hz), −147.09 (2 F, s), −149.33 (2 F, d, 30 Hz), −157.23 (1 F, m, J 10 Hz), −157.38 (2 F, m, J 9 Hz), confirming the Cs symmetry. From the 2D spectrum (not shown) the connectivities were established and all of the peaks identified as shown in the Schlegel diagram (Fig. 4). The spectrum shows the usual characteristics of C60F18 derivatives,15 so that peaks 1 and 3 (which have only one neighbouring sp3-CF group) are downfield, whereas peaks 9 and 10 (which have three such neighbours) are the most upfield and are multiplets.
 | ||
| Fig. 3 19F NMR spectrum for 1. | ||
 | ||
| Fig. 4 Schlegel diagram for 1 (● = F) identifying the fluorine peaks. | ||
This compound is evidently an asymmetrical bis-adduct. The separations in chemical shifts for coupled methylene peaks (0.555, 0.088, 0.898, 0.438 ppm, respectively), together with the far upfield location of one of the doublets, suggests considerable asymmetry; this could result from a methylene hydrogen being close to a fluorine atom, or adjacency of the addends. The most likely combination seems to be addition across both bonds a and b.
This compound is a second unsymmetrical bis-adduct, but the differences in chemical shifts for coupled methylene hydrogens (0.388, 0.306, 0.189, 0.071 ppm) is much smaller than for compound 2. This implies involvement of one of the following combinations (a + b1), (a + b2), (b + b2).
 | ||
| Fig. 5 1H NMR spectrum for 4. | ||
The asymmetry is confirmed by the 19F NMR spectrum (Fig. 6) which comprises eighteen lines (all 1 F intensity) at δF −127.8, −130.35, −134.6, −135.55, −135.8, −136.1, −136.5, −137.45, −137.5, −139.4, −141.05, −142.7, −143.25, −144.35, −149.6, −157.2, −157.95, −138.25. The connectivities were established from the 2D spectrum (inset to Fig. 6) and all of the peaks identified as shown in the Schlegel diagram (Fig. 7). The three upfield peaks are those attached to carbon having three sp3-CF neighbours.
 | ||
| Fig. 6 19F NMR spectrum for 4; inset shows the 2D spectrum with connectivities. | ||
 | ||
| Fig. 7 Schlegel diagram for 4 (● = F) identifying the fluorine peaks. | ||
Resonances for the methylene groups of the symmetrical component 5 consist of two overlapping doublets at δ 4.129 and 4.149 (each 2 H, J 15.6 Hz) and two singlets at δ 3.599 and 3.530 (each 2 H). This can only be obtained from addition across bond a and bond c1 on the opposite side of the fluorinated crown. For the bond c1 addend the hydrogens in each methylene pair are equivalent, so producing singlets. However, one methylene group is much nearer to the fluorinated crown than the other, giving rise to the significant chemical shift of 0.069 ppm. The hydrogens for a given methylene group of the bond a addend are non-equivalent, and so give rise to doublets. Since the addend is further from the fluorines, here there is only a small chemical shift difference of 0.02 ppm. Overall, this result shows that steric hindrance is insufficient to preclude addition across bond c.
The methylene peaks for the unsymmetrical component 6 appear as eight doublets (all 1 H, and ca. 9 Hz), centred at δ 4.04, 3.57; 3.935, 3.57 (two doublets coincide at 3.57 ppm); 3.75, 3.375; 3.55, 3.12. The couplings were determined by NOE. The structure cannot be deduced since there are eight possibilities, but given the identical retention time to that of the symmetrical component, an a + c combination may be involved.
 | ||
| Fig. 8 1H NMR spectrum for 7. | ||
More compelling evidence for the similarities in structures of 1 and 7 is provided by the 19 F NMR spectrum for 7 (Fig. 9), which shows a strong resemblance to that obtained for 1, and consists of ten lines at δF −129.72 (2 F), −135.66 (2 F), −136.78 (2 F), −136.98 (2 F), −141.30 (1 F), −143.11 (2 F, d, J 28 Hz), −148.45 (2 F, d, J 28 Hz), −158.35 (2 F, m), −158.73 (1 F, m). The 2D spectrum was too weak to determine all of the connectivities but they can be deduced (Fig. 10) from intensities, multiplicities, and positions in the 1D spectrum.
 | ||
| Fig. 9 19F NMR spectrum for 7. | ||
 | ||
| Fig. 10 Schlegel diagram for 7 (● = F) identifying the fluorine peaks. | ||
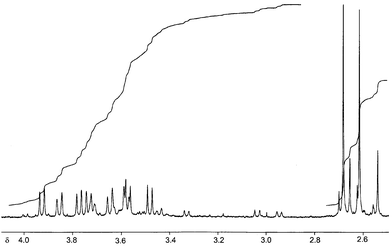 | ||
| Fig. 11 1H NMR spectrum for 9. | ||
The asymmetry is confirmed by the 18-line 19F NMR spectrum (Fig. 12, all 1 F intensity lines and singlets except where indicated) at δF −127.02, −133.88, −134.29, −135.05, −135.77, −136.51 (2 F), −136.71, −137.40 (2 F), −141.64 (d, J 22.5 Hz), −141.21, −143.50 (d, J 22.5 Hz), −148.93 (d, J 26.3 Hz), −151.55 (d, J 26.3 Hz), −157.73 (2 F, m), −158.53 (m). There was insufficient material for a 2D spectrum, so the peaks cannot be assigned. However, the three upfield fluorines are those attached to carbons surrounded by three sp3 CF groups, and one will be adjacent to the most downfield peak. The two pairs of doublets, being relatively upfield will be due to four of the six fluorines that are more remote from the central benzenoid ring.
 | ||
| Fig. 12 19F NMR spectrum for 9. | ||
Both spectra showed the presence of a minor component, which was not identified.
The 19F NMR spectrum was less good than for the other components because of the small sample size, but showed 17 lines (one being of 2 F intensity due to coincidence), at δF −124.04, −133.67, −134.10, −135.06, −135.52 (2 F), −136.08, −136.41, −138.67, −138.99, −139.57, −141.36 (br s), −143.23 (br s), −149.71 (2 F, d, J 24 Hz), −154.20 (2 F, d, J 24 Hz), −156.63, −157.67, −158.54. Again there are three upfield peaks due to fluorines attached to carbons with three sp3 neighbours, one of the three ‘outer’ fluorines gives the peak at −124 ppm (the others cannot be assigned without a 2D spectrum). The four broad singlet and doublet peaks can be assigned to four of the six fluorines that are most remote from the central aromatic ring (see Fig. 1).
Summary of the reaction with formaldehyde
Of the three mono-adducts that can be formed reasonably free of steric hindrance, we have characterised the products arising from addition across bonds a and b, the former being in the largest yield since it is most remote from the fluorines. Given the preponderance of addition across the a bond, the most probable bis-adducts should involve the combinations (a + a1), (a + b), (a + b1), (a + b2), (a + c), (a + c1). Compounds 7 and 5 involve the (a + a1) and (a + c1) combinations, respectively. Compounds 2, 3, and 9 are believed to involve the (a + b), (a + b1), and (a + b2) combinations, with 2 being most probably the former because of the unusual spread of the chemical shifts for the methylene group. Unsymmetrical compound 10 probably involves a combination of (b + c) motifs the adjacency of addition being indicated by the very wide spread of methylene chemical shifts, as for 2 and symmetrical compound 8 must be one of the three possibilities that can result from b,b motif combinations.Compared with reaction on [60]fullerene itself, the presence of the fluorines results in a substantially greater number of possible isomeric products, most of which we have isolated and characterised. In principle it could be possible, by working with larger quantities, to isolate and characterise further components. However, our main objective was to show that the reaction proceeds normally and without loss of fluorine, and further work will now be directed towards minimising bis-addition in reactions with better donor groups. The size of these is likely also to limit the reaction largely to mono-addition across the a bond.
The reaction involving benzaldehyde
This can in principle give many more products than the reaction with formaldehyde. For example, mono-addition across either of bonds a or c can give two products in each case, whilst mono-addition across either of bonds b or d can give four. Steric hindrance in the case of c and d addition would probably reduce these to two and one, respectively, so that the total mono-addition products could number eight. Of the ten fractions obtained (apart from recovered C60F18) those with retention times of 44.6, 26.5, 18.8, and 16.0 min were very minor and were not analysed further.The data show that the minor isomer in each of the 13 and 11.7 min components was the same compound. This could in principle be separated by further HPLC using solvent mixtures but this was unnecessary for our purposes. No CH2 group in any of the three isomers showed any secondary coupling to fluorine in contrast to the other derivatives described below. This shows that the addends in them are remote from fluorine indicating addition across bond a and bond b.
This compound can be either: (i) a mono-addition product involving addition across bond c (the methylene hydrogen adjacent to phenyl being forced towards the fluorines resulting in the observed coupling); (ii) a bis-addition product involving addition across two bonds b. One hydrogen will be nearer to fluorine and so will show the observed secondary coupling. Bis-addition across two bonds a can probably be discounted because neither of the methylene hydrogens should be very near to fluorines. The short retention time would be consistent with a bis-addition product.
Summary of the reaction with benzaldehyde
Six ‘major’ and four minor reaction products were obtained. The former consisted of seven components, one of which is a derivative that was obtained with formaldehyde, three were monoadducts, one could not be resolved, and the remaining two were very probably bis adducts. The amounts of each were insufficient for obtaining 19F NMR spectra, which would facilitate analysis, but this work could be undertaken when larger amounts of C60F18 become available. However, the main purpose of the work at this stage was to verify that the reaction proceeds normally as a preliminary to using more complex addends.The reaction without added aldehyde
To investigate this aspect further, a reaction was carried out with [60]fullerene itself and sarcosine. The normal addition product was obtained having an HPLC retention time of 6 min and giving a parent ion at 777 amu in the mass spectrum. The 1H NMR spectrum showed two singlets at δ 4.35 (CH2) and 2.95 (CH3) identical to the literature value for the normal product obtained from reaction in the presence of formaldehyde.1a A minor component was also present giving δ 4.75 (d, 2 H, J 9.4 Hz, CH2), 4.20 (d, 1 H, J 9.4 Hz), 2.30 (s, 3 H, N–Me), and presumed to be due to bis-additition. Thus formaldehyde is present in the reaction mixture, and to check if the toluene is responsible a further reaction was carried out using sarcosine, benzene and C60F18. Compound 4 was again obtained showing that the formaldehyde is not produced from toluene, so must be present in sarcosine originally, or produced by in situ oxidation by the fullerene.
Acknowledgements
We thank the Royal Society for both a Sino-British Trust Award Fellowship (to X.-W. W.) and a Joint Project Award (to O. V. B. and R. T.).References
- (a) M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798 CrossRef CAS; (b) M. Prato, M. Maggini, C. Giacometti, G. Scorrano, G. Sandonà and G. Farnia, Tetrahedron, 1996, 52, 5221 CrossRef CAS.
- M. Maggini, A. Karlsson, G. Scorrano, G. Sandonà, G. Farnia and M. Prato, J. Chem. Soc., Chem. Commun., 1994, 589 RSC; N. Martin, I. Pérez, L. Sánchez and C. Seoane, J. Org. Chem., 1997, 62, 5690 CrossRef CAS.
- N. Martin, L. Sanchez, C. Seoane, R. Andreu, J. Garin and J. Orduna, Tetrahedron Lett., 1996, 37, 5979 CrossRef CAS.
- M. Maggini, A. Donâ, G. Scorrano and M. Prato, J. Chem. Soc., Chem. Commun., 1995, 845 RSC.
- T. Drovetskaya, C. A. Reed and P. D. W. Boyd, Tetrahedron Lett., 1995, 36, 7971 CrossRef CAS; Y. Sun, T. Drovetskaya, R. D. Bolskar, R. Bau, P. D. W. Boyd and C. A. Reed, J. Org. Chem., 1997, 62, 3642 CrossRef CAS; H. Imahori and Y. Sakata, Chem. Lett., 1996, 199 CAS.
- X. Shi, W. B. Caldwell, K. Chen and C. A. Mirkin, J. Am. Chem. Soc., 1994, 116, 11598 CrossRef CAS.
- F. Novello, M. Prato, T. Da Ros, M. De Amici, A. Bianco, C. Toniolo and M. Maggini, Chem. Commun., 1996, 903 RSC.
- T. De Ros, M. Prato, F. Novello, M. Maggini and E. Banfi, J. Org. Chem., 1996, 61, 9070 CrossRef.
- R. Signorini, M. Zerbetto, M. Meneghetti, R. Bozio, M. Maggini, C. De Faveri, M. Prato and G. Scorrano, Chem. Commun., 1996, 1891 RSC.
- S. R. Wilson, Y. Wang, J. Cao and X. Tan, Tetrahedron Lett., 1996, 37, 775 CrossRef CAS; L. Shu, G. Wang, S. Wu, H. Wu and X. Lao, Tetrahedron Lett., 1995, 36, 3871 CrossRef CAS.
- M. Iyoda, F. Sultana, A. Kato, M. Yoshida, Y. Kuwatani, M. Komatsu and S. Nagase,, Chem. Lett., 1997, 63 CrossRef CAS.
- L. Gan, D. Zhou, C. Luo, H. Tan, C. Huang, M. Lu, J. Pan and Y. Wu, J. Org. Chem., 1996, 61, 1954 CrossRef CAS.
- A. Komori, M. Kubota, T. Ishida, H. Niwa and T. Nogami, Tetrahedron Lett., 1996, 37, 4031 CrossRef CAS.
- Q. Li, D. I. Schuster and S. R. Wilson, J. Org. Chem., 1996, 61, 4764 CrossRef.
- O. V. Boltalina, V. Yu. Markov, R. Taylor and M. P. Waugh, Chem. Commun., 1996, 2549 RSC; A. G. Avent, O. V. Boltalina, P. W. Fowler, A. Yu. Lukonin, V. K. Pavlovich, J. P. B. Sandall, J. M. Street and R. Taylor, J. Chem. Soc., Perkin Trans.2, 1998, 1319 RSC; O. V. Boltalina, B. de La Vaissière, P. W. Fowler, P. B. Hitchcock, J. P. B. Sandall, P. A. Troshin and R. Taylor, Chem. Commun., 2000, 1325 RSC; O. V. Boltalina, B. de La Vaissièrre, P. W. Fowler, A. Yu. Lukonin, J. M. Street and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 2000, 2212 RSC; O. V. Boltalina, P. B. Hitchcock, P. A. Troshin, J. M. Street and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 2000, 2410 RSC; O. V. Boltalina, B. de La Vaissière, A. Yu. Lukonin, P. W. Fowler, A. K. Abdul-Sada, J. M. Street and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 2001, 550 RSC; A. G. Avent, O. V. Boltalina, J. M. Street, R. Taylor and X.-W. Wei, J. Chem. Soc., Perkin Trans. 2, in press. Search PubMed.
- R. Taylor, Lecture Notes on Fullerene Chemistry: A Handbook for Chemists, Imperial College Press, London, 1999, ch. 4. Search PubMed.
- A. D. Darwish, H. W. Kroto, R. Taylor and D. R. M. Walton, Fullerene Science and Technology, 1993, 1, 571 Search PubMed.
| This journal is © The Royal Society of Chemistry 2002 |