Reaction of C60F18 with diethyl bromomalonate: diversion of the Bingel reaction and formation of the first 18π annulenic fullerene†
Received
(in Cambridge, UK)
4th July 2001
, Accepted 26th October 2001
First published on 5th December 2001
Abstract
Reaction of C60F18 with diethyl bromomalonate in the presence of DBU results in the nucleophilic replacement of either one, two, or three of the most accessible fluorine atoms by CBr(CO2Et)2 moieties, in preference to formation of a cyclopropanated derivative (the normal Bingel reaction). Substitution that takes place δ to the departing fluorine, is the first proven example of SN2′ substitution in a fullerene, and appears to be sterically driven. The ratio of mono-/poly-substitution products can be controlled by varying the rate of addition of the DBU and the molar ratio between C60F18 and the other reagents. The tri-substituted product is an [18]annulene, has an intense emerald-green colour ascribable to the electron delocalisation in the (equatorial) annulene belt (bond length variation 0.018 Å), and has C3v symmetry. This
is the first example on an annulenic fullerene (moreover of an all-trans annulene or trannulene). The extent of substitution in each compound is identified from the fluorinated fragments (C60F15, C60F16, and C60F17, respectively, for tri-, di-, and mono-substitution) in the EI mass spectra, and by their 1H and 19F NMR spectra. The structure of the tri-substituted [18]annulene was confirmed by single crystal X-ray diffraction. Normal Bingel cycloaddition also takes place between C60F18 and diethyl malonate–DBU in CBr4, to give C60F18C(CO2Et)2 and C60F16C(CO2Et)2 in relatively low yields. Calculations indicate a critical size of substituent required to produce δ-substitution, rather than ipso-substitution
of the departing fluorine.
The Bingel reaction between diethyl bromomalonate and fullerenes1 is one of the most extensively studied reactions in fullerene chemistry (Scheme 1).2 This, and variations using dialkyl malonates in either the presence or absence of iodine, have been employed for attaching to the cage, via a cyclopropane moiety, donor groups that might give rise to interesting donor–acceptor properties.3 The viability of such derivatives depends upon the strong electron-acceptor properties of the fullerene cage and efforts have been directed towards both increasing the electron supply for the addend, and increasing the electron withdrawal by the cage. However, a ‘catch-22’ situation usually arises with the latter because the sp3 carbons produced by addition, reduce the electron withdrawal. In polyfluorofullerenes there is a
net gain of electron withdrawing power relative to the parent fullerene,4 and we plan to exploit C60F185 in this context because it possesses a substantial ‘curved fullerene’ region6 where cycloadditions may occur. Supplies of C60F18 are presently limited, so initially we are examining some representative reactions to see if the chemistry of the parent fullerenes can be reproduced. In the case of the Bingel reaction we were concerned that the base required for the reaction (DBU) might destroy the fluorofullerene, so that use of this versatile reaction would be precluded.
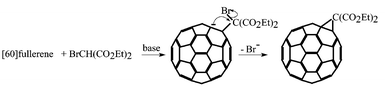 |
| | Scheme 1 Normal course of the Bingel reaction. | |
In the event, these preliminary studies have shown that the reaction takes a different course. After removal of the proton by the base, the resultant nucleophile displaces fluorine from the cage instead of losing bromine to give the normal three-membered ring. This has produced an entirely new type of fullerene derivative which contains a 18π circuit of delocalised electrons, and which we expect to lead to a large family of fullerene derivatives possessing unique spectroscopic properties.
Experimental
An exploratory reaction, carried out by mixing toluene solutions of C60F18 and DBU, was not encouraging. A rapid reaction occurred (colour change), and mass spectrometric analysis of the product showed that all of the fluorine had been lost from the cage. (Ultimately, it may prove possible to use controlled defluorination by DBU to convert polyfluorofullerenes to derivatives of lower and possibly specific addition levels.) Fortunately however, in the presence of diethyl bromomalonate, degradation did not occur and indeed, unreacted C60F18 could be recovered from the mixture, showing that reaction of DBU with the ester was substantially faster than with the fluorofullerene. The reaction produced a mixture of the mono-, di- and tri-substitution products, the amounts varying according to the relative proportions of reagents used, and addition procedure.
In a typical experiment, C60F18 (5 mg) was dissolved in toluene (20 ml, HPLC grade) together with diethyl bromomalonate (1.4 mg dissolved in 1 cm3 of toluene), and DBU (0.85 mg dissolved in 0.4 cm3 of toluene) was added dropwise under argon. The solution turned green immediately, and was stirred at room temperature for a further 1 h. This produced mainly the mono- and tri-substitution product, but a second run using less DBU produced mainly mono- and di-substitution. A third experiment in which the DBU solution was added more slowly to the other reagents produced a greater relative yield of the mono-substitution product. Unreacted C60F18 was recovered for re-use.
In each preparation, a black precipitate formed, and this was removed by filtration, the components of the filtrate being then separated by HPLC (high pressure liquid chromatography). For the initial experiment this employed a Cosmosil 5μ-pye column (10 × 250 mm) with elution by toluene at 4.7 ml min−1. For logistical reasons, subsequent separations employed a 10 × 250 mm Cosmosil Buckyprep column (same flow rate).
For the alternative preparative procedure,7 diethyl malonate (1.14 mg, 1.5 eq.) and CBr4 (2.34 mg, 1.5 eq.) was added to C60F18 (5 mg) in toluene (18 cm3), and then 1.6 eq. of DBU, dissolved in toluene (0.5 cm3), was added slowly. The solution, under argon, was stirred for 5 h, during which time the colour changed from yellow → brown → yellow → green → yellow, and a considerable amount of precipitate was formed. This was removed by filtration and the solution separated by HPLC as before. Identified components eluted at 7.3 and 9.7 min.
Results and discussion
C60F15[CBr(CO2Et)2]3 (1).
HPLC separation of the crude product gave a fraction eluting at 3.55 min/5μ-pye column (2.6 min/Buckyprep column). Re-purification on the Buckyprep column, (elution with 1 ∶ 1 toluene–heptane at 4.7 ml min−1) gave C60F15[CBr(CO2Et)2]3 eluting at 5.0 min. This intense emerald-green compound deposited diamond-shaped crystals from toluene. The spectroscopic properties (1H and 19F NMR, UV–vis, single crystal X-ray diffraction) have been given in a preliminary report.8
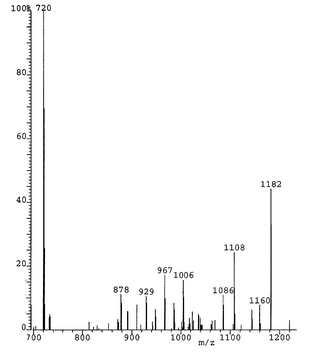
The mass spectrum (Fig. 1S, separately available as supplementary information) showed fragmentation ions m/z at 1639 (M − Br), 1481 [M − CBr(CO2Et)2] and 1005 (C60F15). The latter indicates the number of replaced fluorines (see also the mono- and di-substitution products below). The three-line 19F NMR spectrum indicated C3v symmetry for the product and contrasts markedly with those (below) for the mono- and di-substitution products, and for C60F18 and its derivatives, in lacking the upfield peak that occurs at ca.
−158 ppm when fluorine is attached to carbon surrounded by three fluorinated sp3 carbons.5,9 The fluorine in this location now has only two sp3 neighbours and the signal therefore moves downfield. The 1H NMR spectrum also confirmed
the C3v symmetry.
Two resonance structures for 1 are shown as Schlegel diagrams (Fig. 1) which also includes the equatorial [18]trannulene belt. The two distinct 6 ∶ 5-bonds and the 6 ∶ 6-bond in this belt have lengths of 1.410, 1.397 (nearer to R), and 1.392 Å, respectively, showing extensive resonance delocalisation, the lengths of the corresponding bonds in C60F18 being 1.524, 1.363, and 1.428 Å.6
![Schlegel resonance structures for C60F15[CBr(CO2Et)2]3 (1) showing the delocalised [18]annulene equatorial belt.](/image/article/2002/P2/b105921c/b105921c-f1.gif) |
| | Fig. 1 Schlegel resonance structures for C60F15[CBr(CO2Et)2]3 (1) showing the delocalised [18]annulene equatorial belt. | |
Since all the C–C bonds adjacent to the formal double bonds are trans to one another, this is an all-trans annulene (trannulene),10 the existence of which was conjectured recently.
C60F16[CBr(CO2Et)2]2 (2).
This eluted after 2.8 min (Buckyprep column). Fragmentation ions (m/z) in the mass spectrum (Fig. 2S, separately available as supplementary information) occur at 1479/1481/1483 (M − F), 1261/1263 [M − CBr(CO2Et)2], 1183 [M − CBr(CO2Et)2, Br] and 1024 (C60F16). In contrast to 1, this compound is apple-green in colour (nearer to the lemon yellow–green of C60F18 derivatives) presumably because it does not have a completely conjugated annulene belt.
1H NMR spectrum (Fig. 2).
This is very complex and shows at least nine overlapping methylene quartets between δH 4.58 and 4.37 (2 H, J 7.1 Hz, CH2), due to the eight non-equivalent methylene protons, one of which may be subject to secondary coupling with fluorine at position 1. There are four overlapping equal intensity methyl triplets centred at 1.437, 1.425, 1.414 and 1.411 (3 H, J 7.1 Hz, CH3). The inequivalency in the number of triplets and quartets is attributed to degeneracy in the former. The compound is evidently unsymmetrical and has the structure shown in Fig. 3.
![Schlegel diagram of the structure of C60F16[CBr(CO2Et)2]2 (2); fluorine atom (●) identities were deduced from the 2D 19F NMR spectrum (Fig. 5).](/image/article/2002/P2/b105921c/b105921c-f3.gif) |
| | Fig. 3 Schlegel diagram of the structure of C60F16[CBr(CO2Et)2]2 (2); fluorine atom (●) identities were deduced from the 2D 19F NMR spectrum (Fig. 5). | |
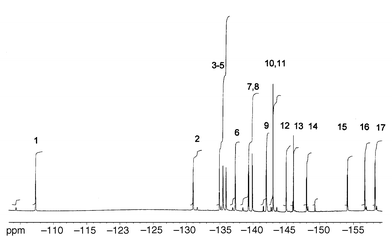
19F NMR spectrum (Fig. 4).
This consists of sixteen lines of equal intensity confirming that two fluorines have been replaced, and that the product is unsymmetrical. From the 2D spectrum (Fig. 5) the fluorine atom locations 1–16 are identified as shown in Fig. 3. Note that in common with other derivatives of C60F18,9 many couplings are evident (ortho, meta, para) across the central benzenoid ring.
C60F17CBr(CO2Et)2 (3).
This mono-substitution product eluted after 6.1 min/5μ pye column or 6.4 min/Buckyprep column. The mass spectrum (Fig. 3S, separately available as supplementary information) shows the bromine-containing parent ion at 1280/1282 amu, and fragment ions at 1201 amu (loss of Br) and 1043 amu (C60F17). The IR spectrum shows main bands at 1157, 1131, 1090, 1069 and 1059 cm−1, cf. 1163, 1133, 1103, 1067 and 1045 cm−1 for C60F18.5 Like the di-substituted derivative (2), this compound also has an apple-green colour.
1H NMR spectrum (Fig. 6).
This shows peaks at δH 4.535 (1 H, dq, J 7.1 and 10.7 Hz), 4.463 (2 H, q, J 7.1 Hz), 4.418, (1 H, dq, J 7.1 and 10.7 Hz), 1.434 (3 H, t, J 7.1 Hz), 1.427 (3 H, t, J 7.1 Hz). As in the case of the di-substituted compound, some triplets are degenerate. The structure is shown in Fig. 7.
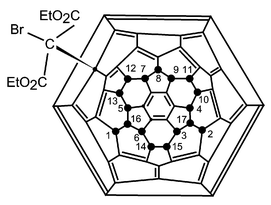 |
| | Fig. 7 Schlegel diagram of the structure of C60F17CBr(CO2Et)2 (3); fluorine atom (●) identities were deduced from the 2D 19F NMR spectrum (Fig. 9). | |
19F NMR spectrum (Fig. 8).
This consists of fifteen lines of equal intensity and one of double intensity confirming that one fluorine has been replaced, and that the product is unsymmetrical. A notable feature is the downfield peak at −107.2 ppm. Peaks in this region are usually found in fluorofullerene spectra only when the C–F bond is adjacent to oxygen, and a through-space interaction between this fluorine atom and one of the oxygens of the ester substituent may be responsible. Moreover, the 2D spectrum (Fig. 9) is unique in showing substantial coupling of this peak to seven others, peaks 5, 6, 10, 13, 14, 15, 16, and presumably the above interaction gives rise to the enhanced coupling. Comparable effects are not seen in the di-substituted derivative (above) possibly because interactions between the substituent arms does not allow one of them to approach the cage so closely. As in the case of the di-substituted compound, some long-range
couplings occur across the central aromatic ring.
UV-vis spectra.
These are shown in Fig. 10 for all three compounds. Because of the small amounts of material currently available and the consequent difficulty of accurate weighing, the extinction coefficients for one compound relative to another are not significant. Above 400 nm, peak maxima occur at ca. 441 (sh), ca. 501 (sh), 548 and 593 nm for the mono-substituted compound 3, at 464, 665 and 735 nm for the di-substituted compound 2, and at 435, 612 and 662 nm for the tri-substituted annulenic compound 1. The intense emerald-green colour of the latter is produced by the band at 662 nm. Both 2 and 3 give shoulder bands in the 380–395 nm region, whereas 1 shows an intense sharp band at 395 nm.
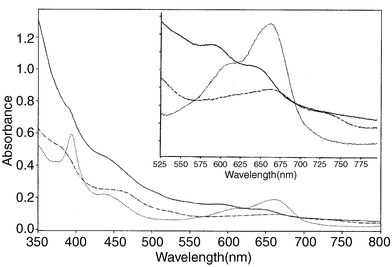 |
| | Fig. 10 UV–vis spectra of 1 (⋯⋯·), 2 (------), and 3 (——); inset shows expansion in the 525–775 nm region. | |
B. Products from the reaction with diethyl malonate
The yields in this reaction were insufficient for full characterisation, but provided enough information to indicate that the expected ‘normal’ Bingel addition takes place under these conditions.
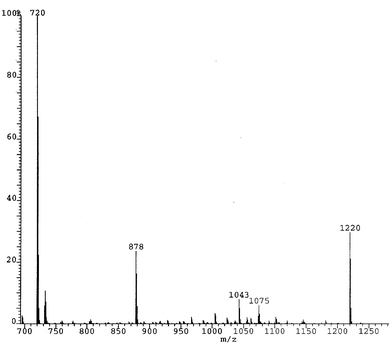
C60F18C(CO2Et)2 (4).
The mass spectrum (Fig. 11) of the fraction eluting after 9.7 min shows the parent ion at 1220 amu corresponding to C60F18C(CO2Et)2. The principal fragmentation ion at 878 amu corresponds to C60C(CO2Et)2 arising from loss of all of the fluorines. There are four possible derivatives that could be obtained by addition of the malonate moiety. In one of these (the most sterically hindered product) the ethyl groups would be equivalent, whereas in the other three they are not. The 1H NMR spectrum shows two quartets centred at δ 4.587 and 4.512 showing that the product is unsymmetrical, which is consistent with addition taking place preferentially across the bond most remote from the fluorines (as happens also in cycloaddition of anthracene).11
C60F16C(CO2Et2)2 (5).
The mass spectrum (Fig. 12) of the fraction that eluted after 7.3 min shows the parent ion at 1182 amu corresponding to C60F16C(CO2Et2)2, with fragmentation down to 878 amu [C60C(CO2Et)2] due to progressive loss of fluorines. It is unlikely that the structure is produced though the malonate moiety substituting two of the fluorines (the product would be very hindered) but rather that the DBU has caused the fluorine loss (a general process confirmed by analysing mixtures of C60F18 and DBU). We have observed loss of two fluorines to give C60F16 under other conditions, and the isolation of the derivative here may arise from C60F16 being also aromatic.12
The substitution process
First we draw attention to our reaction of C60F18 with benzene–FeCl3 which gave C3v symmetry C60F15Ph3 (‘triumphene’), in which the three outermost fluorines of C60F18 were replaced.13 The location of the phenyl groups was inferred by analogy with the formation of C60Ph5Cl from benzene–FeCl3–C60Cl6, in which the five most accessible chlorines are directly substituted by phenyl.14 Given the similar reaction conditions/reagents, a common mechanism for phenyldefluorination was indicated.
In the present instance, the same three fluorines are also replaced, but the incoming nucleophiles occupy positions on the cage different from the departing fluorines. This appears to be sterically driven, with the incoming nucleophile occupying a position δ to the departing fluorine, so that the mechanism is SN2′ (Scheme 2), typical of nucleophilic substitutions when the α-position is hindered. Substitution at the β-position suffers from the same steric compression as at the ipso-position, and is disfavoured with respect to δ-substitution for large incoming groups.
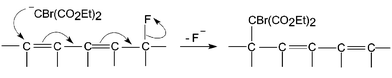 |
| | Scheme 2 Mechanism of replacement of fluorine by the bromomalonyl moiety. | |
The 19F NMR data for C60F15Ph3 gave peaks at δF
−137.3, −138.4, and −145.0, very similar to the above values obtained for the tri-ester (−136.7, −143.9, and −144.0), which could suggest similar structures. The absence in the trannulene of the upfield peak at −158.1 for C60F18 (the one due to fluorine attached to a carbon surrounded by three sp3C–F groups) arises from removal of a fluorine so converting one sp3C–F group to sp2C. In C60F15Ph3 there is likewise no upfield peak in this region, but this corresponds here to the replacement of one fluorine (with its electron-supplying lone pair) by the electron-withdrawing phenyl. Notably, in isostructural C60H1815 the relative positions of the four constituent
peaks are also different to those in C60F18 due to the absence of the lone-pair electron supply from the addend. Thus phenyldefluorination is unlikely to involve δ-substitution, especially since normal substitution by phenyl of an adjacent chlorine occurs in C60Cl6 give C60Ph5Cl, no marked colour was observed, and results of calculations (below).
Semi-empirical molecular orbital calculations of the substitution pathway
In order to rationalise the experimental observations, a series of molecular orbital calculations at the semi-empirical level have been carried out. As a first step the method was calibrated as far as possible, by calculating the energies (AM1) of all C60F18 isomers having the same point group symmetry as that of the isolated and fully characterised isomer (1,2,3,4,8,9,10,16,17,18,22,23,24,36,37,38,39,40-octadecafluoro[60]fullerene).5,6 (For numbering see Fig. 13). This symmetry restriction reduces the many millions of possible isomers to 112, and consideration of only those that converge as closed-shell systems with retention of symmetry reduces the number to below 40, with calculated energies covering a range of over 900 kJ mol−1; it is encouraging that the lowest energy calculated, by 12.5 kJ mol−1, is that for the isomer isolated in experiment.
This ‘crown’ arrangement of the addends has also been identified16 as one of the more stable amongst the C60H18 isomers, and it is known17 that fluorine and hydrogen addends to [60]fullerene give a good structure–energy correlation with each other. The energy of the C60F18 isomer, where the fluorine atoms are at positions 10, 16 and 40 are instead positioned at 30, 44, and 51 lies at over 150 kJ mol−1 above the lowest energy isomer. These are the positions where the new addends are found in 1, i.e. in the δ positions.
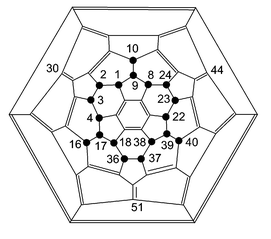 |
| | Fig. 13 Numbering for C60F18. | |
Since the crown structure is the most stable of all the C3v point-group arrangements of addends, it is at first sight surprising that nucleophilic attack does not take place by direct displacement of fluoride ion, i.e. substitution involving frontside attack. The stability of a particular derivative depends more on the arrangement of the addends than on their nature, as the example of fluorine and hydrogen in the previous paragraph shows. However, if the addend is sufficiently large, a new pattern emerges: a good example is the set of structures of the bromo-adducts of [60]fullerene.18 Thus, the next question that calculation may rationalise is: which fluorine atom will be displaced from the original crown structure, and further, which position of the fullerene will be occupied by the incoming group?
The first part of this question may be answered by examination of the charges on the various fluorine atoms and the carbon atoms to which they are attached. Both AM1 and PM3 methods agree in assigning a positive charge of 0.21 to the carbon atom from which the fluoride ion is observed to be lost (10, 16, 40): a charge which is 50% higher than at the next most positively charged carbon. Likewise the ejected fluoride ion is the one bearing the largest negative charge in the original structure. Calculation thus rationalises the experimental observation concerning the identity of the displaced fluorine.
Since a steric effect is the most likely explanation of why the incoming group enters at a position δ to that from which the fluorine is lost (Scheme 2), the energies of a series of C60F15X3 (X = substituent) have been calculated first with X in positions 10, 16, and 40, and then with X in positions 30, 44 and 51. Fifteen fluorine atoms are held in their original positions. The groups X were chosen primarily to give a wide range of steric bulk. The differences in energy between the pairs of isomers are given in Table 1 and plotted against Van der Waals radii in Fig. 14. The correlation is reasonable (r2
= 0.83) and shows that substitution is preferred at the original position until the incoming group is larger than CCl3, when attack at the δ-position becomes favoured. This result suggests
that the phenyl groups in C60F15Ph3 occupy the same positions as the departing fluorines, in support of the structural assignment made in ref. 13.
Table 1 The energy cost of relocating X from positions 10, 16, and 40, to positions 30, 44 and 51 in C60F15X3a
| Substituent X |
Van der Waals radius/pm |
E
rel/kJ mol−1 |
|
Defined as the difference in total energy between the two isomers.
|
| H |
120 |
171 |
| F |
135 |
156 |
| Cl |
180 |
93.3 |
| Ph |
185 |
103 |
| Br |
195 |
66.3 |
| CH3 |
200 |
133 |
| I |
215 |
56 |
| CF3 |
253 |
60 |
| CCl3 |
357 |
23.6 |
| CBr3 |
366 |
−21.5 |
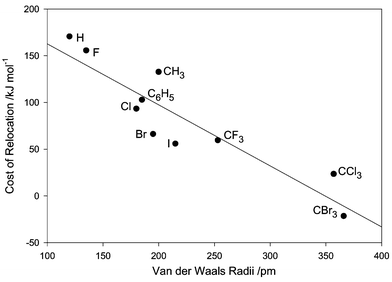 |
| | Fig. 14 Correlation between Van der Waals radii and energy cost of relocation of substituent. | |
Acknowledgements
We thank INTAS (grant no. 97–30027), NATO, and the Royal Society (Joint Project Grant) for financial support (to OVB and RT), the Royal Society for a Sino-British Trust Award Fellowship (to X.-W.W.), and the EU TMR Network USEFULL (Contract FMRX-96–0126).
References
- C. Bingel, Chem. Ber., 1993, 126, 1957 CrossRef CAS; C. Bingel and H. Schiffer, Liebigs Ann. Chem., 1995, 1551 Search PubMed.
- Representative examples are given in
R. Taylor, Lecture Notes on Fullerene Chemistry: A Handbook for Chemists, Imperial College Press, London, 1999, Ch. 9 Search PubMed.
- Recent examples include D. M. Guldi, C. Luo, M. Prato, E. Dietel and A. Hirsch, Chem. Commun., 2000, 373 Search PubMed; M. Diekers, A. Hirsch, C. Luo, D. M. Guldi, K. Bauer and U. Nickel, Org. Lett., 2000, 2, 2741 RSC.
- O. V. Boltalina, L. N. Sidorov, E. V. Sukhanova and I. D. Sorokin, Chem. Phys. Lett., 1994, 230, 567 CrossRef CAS; R. Hettich, C. Jin and R. N. Compton, Int. J. Mass Spectrom. Ion Proc., 1994, 138, 263 CrossRef CAS; F. Zhou, G. J. Van Berkel and B. T. Donovan, J. Am. Chem. Soc., 1994, 116, 5485 CrossRef CAS; N. Liu, H. Touhara, F. Okino and S. Kawasaki, J. Electroanal. Soc., 1996, 143, L214 Search PubMed.
- O. V. Boltalina, V. Yu. Markov, R. Taylor and M. P. Waugh, Chem. Commun., 1996, 2549 RSC.
- I. S. Neretin, K. A. Lyssenko, M. Yu. Antipin, Yu. L. Slovokhotov, O. V. Boltalina, P.
A. Troshin, A. Yu. Lukonin, L. N. Sidorov and R. Taylor, Angew. Chem., Int. Ed., 2000, 39, 3273 CrossRef CAS.
- X. Camps and A. Hirsch, Chem. Commun., 1997, 1595 RSC.
- X.-W. Wei, A. D. Darwish, O. V. Boltalina, P. B. Hitchcock, J. M. Street and R. Taylor, Angew. Chem. Int. Ed., 2001, 40, 2989 CrossRef CAS.
- O. V. Boltalina, B. de La Vaissière, P. W. Fowler, A. Yu. Lukonin, J. M. Street and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 2000, 2215 Search PubMed; O. V. Boltalina, P. B. Hitchcock, P. A. Troshin, J. M. Street and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 2000, 2410 RSC; O. V. Boltalina, B. de La Vaissière, A. Yu. Lukonin, P. W. Fowler, A. K. Abdul-Sada, J. M. Street and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 2001, 550 RSC.
- A. F. Fokin, H. Jiao and P. von R. Schleyer, J.
Am. Chem. Soc., 1998, 120, 9364 CrossRef CAS.
-
A. G. Avent, O. V. Boltalina, J. M. Street, R. Taylor and X.-W. Wei, J. Chem. Soc., Perkin Trans. 2, 2001, 994 Search PubMed.
- A. G. Avent, O. V. Boltalina, A. Yu. Lukonin, J. M. Street and R. Taylor, J. Chem. Soc., Perkin
Trans. 2, 2000, 1359 RSC.
- O. V. Boltalina, J. M. Street and R. Taylor, Chem. Commun., 1998, 1827 RSC.
- A. G. Avent, P. R. Birkett, J. D. Crane, A. D. Darwish, G. J. Langley, H. W. Kroto, R. Taylor and D. R. M. Walton, J. Chem. Soc., Chem. Commun., 1994, 1463 RSC.
- A. D. Darwish, A. G. Avent, R. Taylor and D. R. M. Walton, J. Chem. Soc., Perkin Trans. 2, 1996, 2051 RSC.
- B. W. Clare and D. L. Kepert, J. Mol. Struct. (THEOCHEM), 1994, 303, 1 CrossRef.
- O. V. Boltalina, M. Bühl, A. Khong, M. Saunders, J. M. Street and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1999, 1475 RSC; P. W. Fowler and J. P. B. Sandall, J. Chem. Soc., Perkin Trans. 2, 1997, 419 RSC.
- P. R. Birkett, P. B. Hitchcock, H. W. Kroto, R. Taylor and D. R. M. Walton, Nature, 1992, 357, 479 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2002 |
Click here to see how this site uses Cookies. View our privacy policy here. ![1H NMR spectrum for C60F16[CBr(CO2Et)2]2 (2).](/image/article/2002/P2/b105921c/b105921c-f2.gif)
![19F NMR spectrum (376.4 MHz) for C60F16[CBr(CO2Et)2]2 (2).](/image/article/2002/P2/b105921c/b105921c-f4.gif)
![2 D 19F NMR spectrum for C60F16[CBr(CO2Et)2]2 (2).](/image/article/2002/P2/b105921c/b105921c-f5.gif)