Isolation and characterisation of symmetrical C60Me6, C60Me5Cl and C60Me5O2OH, together with unsymmetrical C60Me5O3H, C60Me5OOH, C60Me4PhO2OH, and C60Me12; fragmentation of methylfullerenols to C58
Hamad
Al-Matar
a,
Ala'a K.
Abdul-Sada
a,
Anthony G.
Avent
a,
Patrick W.
Fowler
b,
Peter B.
Hitchcock
a,
Kevin M.
Rogers
b and
Roger
Taylor
*a
aThe Chemistry Laboratory, School of Chemistry, Physics and Environmental Sciences, University of Sussex, Brighton, UK BN1 9QJ
bSchool of Chemistry, University of Exeter, Exeter, UK EX4 4QD
First published on 4th December 2001
Abstract
Reaction of freshly prepared C60Cl6 (from chlorination of [60]fullerene by ICl in benzene) with methyllithium followed by hydrolysis and work-up including HPLC separation yields Cs symmetry C60Me6 (isostructural with C60Br6 and C60Cl6), together with unsymmetrical C60Me12 which is comprised of two of the motifs present in C60Me6 and must arise from the presence of a small amount of C60Cl12 in the C60Cl6. From the same reaction mixture we have also obtained C60Me5Cl [isostructural with C60Ar5Cl and C60(OR)5Cl], hydroxyepoxides [C60Me5O2OH (symmetrical), C60Me5OOH and C60Me4PhO2OH (both unsymmetrical)] and unsymmetrical C60Me5O3H (a cage-opened ketone). The results provide further information concerning the addition patterns and mechanistic features of fullerene chemistry, show that methylated, arylated, alkoxylated and halogenated [60]fullerenes are isostructural, and that C60Cl6 also contains traces of C60PhCl5. Some of the compounds give exceptionally high intensities of the C58+ fragmentation ion during EI mass spectrometry.
Introduction
The great majority of fullerene chemistry studies concern cycloadditions. They are relatively easy to carry out, and a single major product can usually be obtained in good yield. Steric hindrance due to the 1,2-cycloaddend inhibits subsequent reaction at the 3,4-bond, the site otherwise preferred since it has enhanced π-density created by the first addition.1,2Reactions involving polyaddition have been less studied because control of the addition level is difficult, and further reaction in the vicinity of the first addition also occurs. Nevertheless, study of these reactions is essential for fundamental understanding of the electronic and steric effects that operate in the cage. Structural analysis is especially difficult when the products have unsymmetrical arrays, since there is no analytical technique presently available (unless suitable and ordered crystals are obtained) for determining the addend dispositions. In these cases deductive reasoning based on NMR data, and intuition (with the possibility of error) has to be resorted to in the interim.
Hydrogenation is at first sight the ideal reaction of choice, since steric effects should be minimal, and some studies aimed at locating the addends in this reaction have been carried out.2–6 However, hydrogenation is complicated both by the tendency of derivatives to oxidise rapidly to the fullerenol, and multiple spin–spin coupling of higher hydrogenated species which prevents interpretation of some NMR spectra. We have therefore investigated alkylation, since alkylfullerenes are comparatively stable, give analysable NMR spectra, and dissolve readily in solvents in which the parent fullerene is virtually insoluble.
Alkylation requires in principle, electrophilic addition, a process rendered difficult by the electron-withdrawing properties of the cage. This can be circumvented either by reaction of the electrophile with the fullerene radical anion (produced both by reaction with alkali metals), or by nucleophilic substitution of a halogeno group by alkyl; we have used the latter method in this study. The former, introduced by Olah and co-workers, resulted in the addition of up to 24 methyl groups to the cage, with (uncharacterised) C60Me6 and C60Me8 prominent.7 More controlled addition can be obtained by initial electrochemical reduction, and in this way a mixture of 1,2- and 1,4-C60Me2 has been obtained,8 and also C60R2 (R = Et, n-Bu), C60Me4 and C60Me6, (all so far uncharacterised).9 In the sublimate from the reaction of [60]fullerene with potassium–MeI, C60Me6 (uncharacterised) was the main product.10
An alternative means of introducing alkyl groups onto [60]fullerene involves addition of nucleophilic alkyl from reagents MR (M = alkali metal), with quenching of the intermediate C60Rn− either by electrophilic H or alkyl R′.11 This has been used to produce C60R5H (R = fluoren-9-yl),12 which is isostructural with C60Ph5H,13 and a number of other derivatives (many as yet uncharacterised).11 A related procedure giving C60Me5H employed the organocopper reagent mixture, MeMgBr–CuBr–Me2S.14 Alkylation has also been observed within a mass spectrometer by reacting fullerenes with ketones (up to ca. 20 alkyl groups become attached),15 and cyanoalkyl groups have been attached to [60]fullerene by reaction with azoisobutyronitrile.16
Our previous study on methylation through reaction between both [60]- and [70]fullerenes with lithium followed by methyl iodide, revealed the following.17
(i) The detection and/or identification of various methylated [60]fullerene species viz., 1,2- and 1,4-Me2C60; unsymmetrical Me6C60 (showing three NOE pairs of methyls); six different isomers of Me4C60 (each of which has either Cs or C2 symmetry); Me8C60 [which has the C2v structure motif shown in 1, found previously only in C60Br8].18

(ii) Up to 34 methyl groups added to [60]fullerene (this confirms the general observation of Olah and coworkers),7 the most abundant species (EI mass spectrum) in the polymethylated mixture being C60Men (n = 10, 12, 14). These results are pertinent to recent calculations which indicate that [60]fullerene can accommodate 12 electrons in Li12C60, but no further electrons are transferred to the cage at higher lithiation levels.19 Thus lithiation followed by reaction with methyl iodide should give C60Me12 as the maximum methylation level. Since much higher levels are observed in the reaction of [60]fullerene with lithium metal in solution, either the calculations do not model the experiments, or an extremely rapid series of consecutive reactions occur viz., quenching → further lithiation → quenching → further lithiation. Given the instantaneous reaction that occurs on quenching with methyl iodide, further lithiation at this stage seems highly improbable. Fragmentation–recombination of lower methylated species during EI mass spectrometry may be discounted since we were able to obtain spectra of pure methylfullerenes free from any higher methylated species.
(iii) 1,2-Me2C60 readily undergoes atmospheric oxidation to give five different oxide derivatives.
(iv) [70]Fullerene adds mainly 2, 4, 6, 8, or 10 methyl groups, and 1,2- and 5,6-Me2C70 are produced in a 3.4 ∶ 1 ratio.
(v) Reduction of 1,2- and 1,4-Me2C60 takes place to give either Me2C60H16 or Me2C60H34, i.e. 18 or 36 overall addend levels, emphasising the fundamental significance of these levels, which have been observed in hydrogenation and fluorination.3,20
(vi) The solubility increases with extent of methylation, and becomes high in solvents such as acetone and THF in which the fullerenes themselves are insoluble.
(vii) The retention times on a Cosmosil Buckyprep column decrease with increased methylation level. (NB The retention times of 6.2 and 6.5 min given for 1,2- and 1,4-Me2C60, respectively, in ref. 1 should be interchanged.)
We now describe our results obtained from reaction of C60Cl6 with methyllithium.
Experimental
C60Cl6 was prepared by chlorinating [60]fullerene with ICl as described previously,21 and was used without further purification in order to avoid degradation.An excess of a MeLi solution (4 ml of 1 M in THF–cumene, 11 ∶ 9) was stirred under N2 with C60Cl6 (100 mg) at room temp. The orange solution turned brown–black immediately, and stirring was continued overnight. The reaction mixture was extracted with toluene, washed with water, dried (MgSO4), and the solvent removed under vacuum. Column chromatography (70–230 mesh silica gel), gave after elution with cyclohexane–toluene (9 ∶ 1) a major fraction which contained C60Me6 and C60Me5Cl. Further elution with cyclohexane–toluene (1 ∶ 1) yielded symmetrical C60Me5O2OH, and finally, elution with toluene alone gave a mixture of (all unsymmetrical) C60Me5O3H, C60Me5OOH, and C60Me4PhO2OH.
HPLC separation of the products was carried out using a 10 mm × 250 mm Cosmosil ‘Buckyprep’ column operated at a flow rate of 4 ml min−1, with elution either by toluene or toluene–heptane, (1 ∶ 1 v/v). The retention times accompany details of the isolated components, below.
All EI mass spectra were run at 70 eV. 1H NMR spectra were run as solutions in CDCl3, and IR spectra were obtained using KBr discs.
Results and discussion
(i) C60Me5Cl
This compound (4 mg, 4.5%) eluted after 4.9 min (toluene) or 9.8 min (1 ∶ 1 toluene–heptane). In the EI mass spectrum (Fig. 1) the parent ion at 830/832 amu is just discernible, but due to the fragmentation which chlorofullerenes readily undergo under EI conditions, the main peak at 796 amu is due to chlorine loss followed by hydrogen capture; the subsequent fragmentation ions arise from consecutive loss of five methyl groups. In the doubly charged region, peaks are seen only for the fragmentation ions. | ||
| Fig. 1 EI mass spectrum for C60Me5Cl. | ||
The IR spectrum exhibits the C–H stretching frequencies for the methyl groups at 2959, 2925 and 2854 cm−1.
The 1H NMR spectrum (Fig. 2) shows three methyl groups at δ 2.56, 2.355 and 2.335 in a 1 ∶ 2 ∶ 2 intensity ratio (the peak × at δ 2.365 is due to traces of toluene), hence the compound has Cs symmetry. These shifts may be compared to those for the corresponding C60Me5H which are δ 2.42, 2.32, 2.30.14 The downfield shift of the single methyl group resonance relative to those for the other methyls and relative to C60Me5H show that it must be next to the electronegative chlorine. Further, there are 0.3% and 0.7% NOE enhancements between the A and B methyls but none between the A and C methyls, confirming that the compound (Fig. 3) is isostructural with C60Ar5Cl,13 and with C60(OR)5Cl (R = Me, Et).22 As in these latter cases, the least accessible chlorine is less readily replaced than the others.
 | ||
| Fig. 2 1H NMR spectrum for C60Me5Cl, with peaks identified as shown in Fig. 3. | ||
 | ||
| Fig. 3 Structure of C60Me5Cl. | ||
(ii) C60Me6
This compound (6 mg, 7.3%) eluted after 4.4 min (toluene) or 8.1 min (1 ∶ 1 toluene–heptane). and gave an excellent EI mass spectrum (Fig. 4), which furthermore exhibits alternation in peak intensities due to consecutive loss of methyl groups, analogous to that found with phenylated fullerenes.23 The 1H NMR spectrum shows four methyl peaks at δ 2.364, 2.300, 2.281 and 2.264 in a 1 ∶ 2 ∶ 2 ∶ 1 ratio showing that the molecule has Cs symmetry. The NOE couplings of 1.3 and 1.8% between the A and D methyls confirm the structure as shown in Fig. 5, the methyl groups occupying the 1, 2, 4, 11, 15, and 30-positions. The compound (the IUPAC name24 of which is 1,2,4,11,15,30-hexamethyl-1,2,4,11,15,30-hexahydro[60]fullerene) is thus isostructural with C60allyl6,25 the only other hexaalkyl[60]fullerene characterised to date.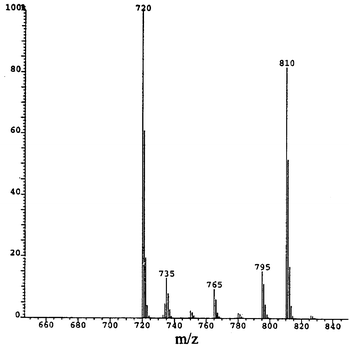 | ||
| Fig. 4 EI mass spectrum for C60Me6. | ||
 | ||
| Fig. 5 Structure of C60Me6. | ||
The structure of the compound was confirmed by the single crystal X-ray structure (Fig. 6, 20% ellipsoids) obtained from crystals grown from toluene. The C(cage)–Me bond lengths (in Å) are 1.575 (C2–C66), 1.552 (C1–C61), 1.536 (average of C4–C62 and C11–C65) and 1.530 (average of C15–C63 and C30–C64). Thus steric compression causes significant bond lengthening when the methyl groups are adjacent, this being greatest for the C2-methyl group. Elongation of the bond to C2 was also observed previously in isostructural C60Br6.18
 | ||
| Fig. 6 Single crystal X-ray structure of C60Me6. | ||
(iii) Symmetrical C60Me5O2OH
This compound (7 mg, 8%) eluted after 4.8 min (toluene) or 8.8 min (1 ∶ 1 toluene–heptane), and the structure (Fig. 7) has been fully characterised in a preliminary publication.26 [Owing to a lock signal error, the reported 1H NMR resonances should each be downfield by 0.37 ppm, i.e. at δ 4.25 (OH, confirmed by saturation transfer to water), 2.36 (MeA), 2.23 (MeC), and 2.12 (MeB).]; IR/cm−1 3520br, 2971, 2924, 2857, 1438, 1384, 1099, 1074, 1047, 1037, 1016, 941, 665, 658, 572, 553, 535 and 513. | ||
| Fig. 7 Structure of symmetrical C60Me5O2OH. | ||
A notable feature to which we draw attention here is that the C58 fragmentation ion at 696 amu in the EI mass spectrum is 40% of the intensity of the 720 amu peak (see also below). This intensity is very much higher than is found in the EI mass spectra of C60 and arises because of the more facile loss of 2 CO molecules. We have noted this previously in the mass spectra of phenylated epoxides of [60]fullerene, where the intensity of the 696 amu peak was 30% of that of the 720 amu peak;27 even higher intensities are found with unsymmetrical C60Me5O3H and C60Me4PhOOH (below).
(iv) Unsymmetrical C60Me5O3H
This compound (4 mg, 5%), which eluted after 5.2 min (toluene) or 10.1 min (1 ∶ 1 toluene–heptane), is an open-cage ketone, and full details of the structural analysis have been described.28 The intensity of the C58+ fragmentation ion (696 amu) in the EI mass spectrum was 55% of that of the 720 amu peak.(v) C60Me5OOH
This compound (7 mg, 8%) eluted after 4.9 min (toluene) and 9.9 min (1 ∶ 1 toluene–heptane). The EI mass spectrum (Fig. 8) shows the parent ion at 828 amu; here the intensity of C58+ relative to that of C60 arising from 2 × CO loss is also substantial (28%). IR/cm−1 3492, 2963, 2921, 2859, 1443, 1417, 1377, 1342, 1267, 1238, 1201, 1163, 1104, 1068, 1028, 1015, 925, 733, 684, 661, 576, 553, 529 and 507. | ||
| Fig. 8 EI mass spectrum for C60Me5OOH. | ||
The 1H NMR spectrum (Fig. 9) shows peaks at δ 3.59 (1 H, s, OH), 2.36 (3 H, s, MeA), 2.30 (3 H, s, MeC), 2.24 (3 H, s, MeC′), 2.22 (3 H, s, MeB), 2.14 (3 H, s, MeB′); the identity of the OH group was confirmed by saturation transfer to water. The compound is therefore unsymmetrical, the methyl peak locations being very similar to those in symmetrical C60Me5O2OH. These peaks are identified from the NOE couplings which are 0.3, 0.2, 0.2, 0.1, and 0.2%, between OH and MeA, MeB, MeB′, MeC, and MeC′, respectively, and 2.7, 1.5, 2.4, 0.7, and 0.6% respectively between the methyls and OH.
 | ||
| Fig. 9 1H NMR spectrum for C60Me5OOH. | ||
The 13C NMR spectrum shows the required 52 peaks for the cage sp2-carbons at δC 156.93, 154.88, 152.85, 152.18, 152.13, 150.58, 149.16, 148.85, 148.64, 148.62, 148.44, 148.38, 148.35, 148.32, 148.27 (2 C), 148.26, 148.22, 148.19, 148.10, 147.95, 147.69, 147.53, 147.525, 147.39, 147.29, 147.16, 147.025, 146.40, 146.16, 144.41, 144.29, 145.93, 144.55, 144.20, 144.18, 144.12, 144.06, 143.81, 143.80, 143.56 (3 C), 143.49, 143.35, 143.24, 143.00, 142.97, 142.68, 142.59, 142.47, 139.22. In the sp3 region peaks appear at δC 84.00, 80.76, and 75.95 (all 1 C, due to C–O–C and C–OH), 52.60 (C–MeA), 50.50 (C–MeC), 50.43 (C–MeC′), 47.30 (C–MeB), 46.28 (C–MeB′), 27.75 (MeA), 25.63 (MeC), 25.17 (MeC′), 24.71 (MeB), 23.87 (MeB′).
Single crystals produced only weak diffraction and showed two independent molecules with, in both cases, the oxygen atoms disordered. Results were consistent with the structure in Fig. 10 deduced from the other data.
 | ||
| Fig. 10 Structure of C60Me5OOH. | ||
An interesting feature of this compound that it is isostructural with C60Ph5O2H, a species which undergoes oxidative dehydrogenation to C60Ph4C6H4O2 (which contains a furanoid ring).29 A comparable oxidation is unlikely in the present case because it would lead to formation of a very strained four-membered ring.
(vi) C60Me4PhO2OH
This compound (4 mg, 5%) eluted after 4.9 min (toluene) or 10.7 min (1 ∶ 1 toluene–heptane). The EI mass spectrum (Fig. 11) shows the parent ion at 906 amu and has a very intense C58+ peak (696 amu), which is 76% of that for C60. This is much the highest relative intensity so far observed for this ion in any fullerene derivative. | ||
| Fig. 11 EI mass spectrum for C60Me4PhO2OH. | ||
The 1H NMR spectrum gave δ 7.80–7.78 (2 H, dm, J 7.2 and 0.7 Hz), 7.52–7.48 (2 H, dt, J 7.2 and 0.7 Hz), 7.42–7.38 (1 H, dt, J 7.2 Hz and unresolved), 4.285 (1 H, OH), 2.48 (3 H, s, Me), 2.175 (3 H, s, Me), 2.171 (3 H, s, Me), 2.095 (3 H, s, Me). The locations of the addends were deduced initially from the NOE couplings [Fig. 12(a)] and confirmed later by the single crystal X-ray structure [Fig. 12(b), 20% ellipsoids].
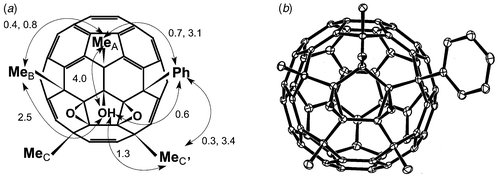 | ||
| Fig. 12 C60Me4PhO2OH showing: (a) NOE couplings (%); (b) single crystal X-ray structure. | ||
The question arises as to the origin of the phenyl group. Chlorination of [60]fullerene to give C60Cl6 is carried out with ICl in benzene solution, and HPLC analysis of the product30 shows that by-products comprise as much as 25% of the total yield. These have not been characterised because of the ready elimination of chlorine during EI mass spectrometry, but probably contain various combinations of phenyl and chloro addends, produced by electrophilic substitution into the benzene solvent; the high electrophilicity of the cage allows this to occur even in the absence of Friedel–Craft catalysts. We may assume that one of these will be C60Cl5Ph, so giving rise to the observed derivative, but this does not explain the location of the phenyl group. Whilst occupation of the MeA position by Ph would probably increase steric hindrance, this is not the case for occupation of the MeC′ position. Possibly, other isomers are formed which we have not isolated.
(vii) C60Me12
The 1H NMR spectrum of a fore-run of C60Me6 shows also the presence of eleven other peaks (ten of equal intensity and one of double intensity) in the lower field region, at δ 2.196, 2.172, 2.149, 2.146, 2.075, 2.057, 2.007, 1.997, 1.927, 1.920, 1.840 (2 H) (Fig. 13). There are NOE enhancements of 2.0% between the δ 2.196 and 2.172 peak pair and 1.8% between the δ 1.927 and 1.920 peak pair. The higher the addition level, the further upfield are the peaks in the 1H NMR spectra of fullerenes (see e.g. ref. 3), so this by-product is evidently unsymmetrical C60Me12. From the peak integration, the C60Me6 ∶ C60Me12 ratio is 55 ∶ 45. The two NOE couplings indicate that C60Me12 contains two of the motifs shown in Fig. 5. There are twelve ways in which these two arrangements can be combined, nine with adjacent motifs differing only in the relative positions of the addends attached to the central pentagons (1 × C2v, 2 × Cs, 2 × C2, 4 × C1) and three with remote motifs, centred on antipodal pentagons of the cage (1 × C2h, 2 × C2). With a variety of addends (H, F, Cl, Br, Me) and all three MO semiempirical methods, the three antipodal isomers are consistently more stable than the adjacent isomers by 20–30 kJ mol−1. However, their formation would not be consistent with contiguous addition31 and all three are ruled out by symmetry. The number of peaks in the spectrum is consistent only with the four totally unsymmetrical C1 isomers with adjacent motifs. These have addends at the following positions (see ref. 32 for numbering) 1, 2, 4, 7, 11, 15, 20, 22, 23, 30, 37, 40 (No. 3); 1, 2, 4, 11, 15, 18, 30, 34, 35, 38, 51, 54 (No. 4); 1, 2, 4, 7, 11, 15, 19, 23, 30, 37, 39, 40 (No. 5); 1, 2, 4, 11, 15, 18, 30, 34, 38, 51, 53, 54 (No. 8). There is no obvious mechanistic reason for any one of these to be preferred, and the semiempirical calculations actually favour more symmetric adjacent-motif isomers over all four. In kinetic models based on Hückel theory all nine adjacent-motif isomers would have equal energy and the product would be a statistical mixture. In MOPAC calculations, MNDO and AM1 methods prefer isomer No. 8 amongst the C1 set by a margin of 5–18 kJ mol−1, but PM3 prefers isomer No. 4 by a margin of 1–6 kJ mol−1. No further conclusions regarding the structure of C60Me12 can be made, short of obtaining a single-crystal X-ray structure. | ||
| Fig. 13 1H NMR spectrum of C60Me12. | ||
Solubility of methylfullerenes
The solubilities of a mixture of methylated [60]fullerenes obtained by methylation with lithium–MeI are (mg ml−1): THF (30), acetone (3), dichloromethane (3), CS2 (6), toluene (3), benzene (3), slight solubility being observed in petroleum ether, cyclohexane and heptane. The THF and acetone solubilities are orders of magnitude greater than that of [60]fullerene, and suggest applications of these derivatives (e.g. as polymer cross-linkers) in which the parent fullerenes show promising properties, but insufficient solubility.33Crystal data for C60Me6†
M = 810.8, monoclinic, P21/n (No. 14), a = 11.6277(6), b = 19.6831(10), c = 15.1972(8) Å, α = 90, β = 100.541(3), γ = 90°, V = 3419.5(3) Å3, Z = 4, μ(Mo-Kα) = 0.09 mm−1, T = 173 K. Rf = 0.123 for 4271 reflections with I > 2σ(I), wR2 = 0.334 for 5932 independent reflections.Crystal data for C60Me4PhO2OH†
M = 906.84, monoclinic, P21/n (No. 14), a = 10.0491(2), b = 31.5976(8), c = 11.6546(3) Å, α = 90, β = 93.197(1), γ = 90°, V = 3694.9(2) Å3, Z = 4, μ(Mo-Kα) = 0.10 mm−1, T = 173 K. Rf = 0.045 for 3691 reflections with I > 2σ(I), wR2 = 0.1058 for 4462 independent reflections. The O atoms are disordered 0.69 ∶ 0.31 over two arrangements (O1 at C1 or C3).Acknowledgements
We thank the University of Kuwait for a research grant (to H. A.-M.).References
-
R. Taylor, Lecture Notes on Fullerene Chemistry: A Handbook for Chemists, Imperial College Press, London, 1999, chs. 3 and 9 Search PubMed
.
- A. G. Avent, A. D. Darwish, D. K. Heimbach, H. W. Kroto, M. F. Meidine, J. P. Parsons, C. Remars, R. Roers, O. Ohashi, R. Taylor and D. R. M. Walton, J. Chem. Soc., Perkin Trans. 2, 1994, 15 RSC
- A. D. Darwish, A. G. Avent, R. Taylor and D. R. M. Walton, J. Chem. Soc., Perkin Trans. 2, 1996, 2051 RSC
- C. C. Henderson, C. M. Rohlfing and P. A. Cahill, Chem. Phys. Lett., 1992, 213, 383 CrossRef CAS
- M. S. Meier, P. S. Corbin, V. K. Vance, M. Clayton, M. Mollman and M. Poplawska, Tetrahedron Lett., 1994, 32, 5789 CrossRef CAS
- W. E. Billups, L. Luo, A. Gonzalez, D. Arguello, L. B. Alemay, T. Marriott, M. Saunders, H. A. Jimenez-Vázquez and A. Khong, Tetrahedron Lett., 1997, 38, 171, 175
- J. W. Bausch, G. K. S. Prakash, G. A. Olah, D. S. Tse, D. C. Lorents, Y. K. Bae and R. Mahlhotra, J. Am. Chem. Soc., 1991, 113, 3205 CrossRef CAS
- C. Caron, R. Subramanian, F. D'Souza, J. Kim, W. Kutner, M. T. Jones and K. M. Kadish, J. Am. Chem. Soc., 1993, 115, 8505 CrossRef CAS
- K. M. Kadish, personal communication.
- G. P. Miller, personal communication.
-
R. Taylor, Lecture Notes on Fullerene Chemistry: A Handbook for Chemists, Imperial College Press, London, 1999, ch.
6 Search PubMed
- Y. Murata, K. Komatsu and T. S. M. Wan, Tetrahedron Lett., 1996, 37, 7061 CrossRef CAS
- A. G. Avent, P. R. Birkett, J. D. Crane, A. D. Darwish, G.
J. Langley, H. W. Kroto, R. Taylor and D. R. M. Walton, J. Chem. Soc., Chem. Commun., 1994, 1463 RSC
- M. Sawamura, M. Toganoh, Y. Kuminobu, S. Kato and E. Nakamura, Chem. Lett., 2000, 270 CrossRef CAS
- E. A. Shilova, Y. I. Lyakhovetsky, B. L. Tumanskii, A. I. Belokon and Y. S. Nekrasov, Mendeleev Commun., 1999, 176 CrossRef
- W. T. Ford, T. Nishioka, F. Qiu, F. D'Souza, J. Choi, W. Kutner and K. Noworyta, J. Org. Chem., 1999, 64, 6257 CrossRef CAS
- H. Al-Matar and R. Taylor, Rec.
Adv. Chem. Phys. Fullerenes Relat. Mater., 1999, 7, 163 Search PubMed
- P. R. Birkett, P. B. Hitchcock, H. W. Kroto, R. Taylor and D. R. M. Walton, Nature (London), 1992, 357, 479 CrossRef CAS
- J. Kohanoff, W. Andreoni and M. Parrinello, Chem. Phys. Lett., 1992, 198, 472 CrossRef CAS
- A. D. Darwish, A. K. Abdul-Sada, G. J. Langley, H. W. Kroto, R. Taylor and D. R. M. Walton, J. Chem. Soc., Perkin Trans. 2, 1995, 2359 RSC
- P. R. Birkett, A. G. Avent, A. D. Darwish, H. W. Kroto, R. Taylor and D. R. M. Walton, J. Chem. Soc., Chem. Commun., 1993, 1230 RSC
- A. G. Avent, P. R. Birkett, A. D. Darwish, S. Houlton, R. Taylor, K. S. T. Thomson and X.-W. Wei, J. Chem. Soc., Perkin Trans. 2, 2001, 782 RSC
- P. R. Birkett, A. G. Avent, A. D. Darwish, H. W. Kroto, R. Taylor and D. R. M. Walton, Fullerene Sci. Technol., 1997, 5, 705 CAS
- E. W. Godly and R. Taylor, Pure Appl. Chem., 1997, 69, 1411 CAS
- A. K. Abdul-Sada, A. G. Avent, P. R. Birkett, H. W. Kroto, R. Taylor and D. R. M. Walton, J. Chem. Soc., Perkin Trans. 1, 1998, 393 RSC
- H. Al-Matar, P. B. Hitchcock, A. G. Avent and R. Taylor, Chem. Commun., 2000, 1071 RSC
- A. D. Darwish, P. R. Birkett, G. J. Langley, H. W. Kroto, R. Taylor and D. R. M. Walton, Fullerene Sci. Technol., 1997, 5, 705 CAS
- H. Al-Matar, A. K. Abdul-Sada, A. G. Avent and R. Taylor, Org. Lett., 2001, 3, 1669 CrossRef CAS
- A. D. Darwish, A. G. Avent, H. W. Kroto, R. Taylor and D. R. M. Walton, Chem. Commun., 1997, 1579 RSC
- A. D. Darwish and R. Taylor, unpublished work.
- A. G. Avent, A. D. Darwish, D. K. Heimbach, H. W. Kroto, M. F. Meidine, J. P. Parsons, C. Remars, O. Ohashi, R. Roers, R. Taylor and D. R. M. Walton, J. Chem. Soc., Perkin Trans. 2, 1994, 15 RSC
-
R. Taylor, Lecture Notes on Fullerene Chemistry: A Handbook for Chemists, Imperial College Press, London, 1999, ch. 2 Search PubMed
- Y. Wang, N. Herron, R. V. Kasowski, A. Suna and K.-S. Lee, The Chemical Physics of Fullerene 10 (and 5) Years Later,
(NATO ASI Ser., Ser. E), 1996, 316, 329 Search PubMed
Footnote |
| † Full structural data are available from the Cambridge Crystallographic Data Centre (CCDC 166355 and 166356). See http://www.rsc.org/suppdata/p2/b1/b108292m/ for crystallographic files in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2002 |