Pyrido[1,2-a]quinoxalines: synthesis, crystal structure determination and pH-dependent fluorescence†
Received
(in Cambridge, UK)
26th March 2001
, Accepted 31st October 2001
First published on 3rd December 2001
Abstract
A series of novel, fluorescent pyrido[1,2-a]quinoxalines have been synthesized with crystal structures obtained for several analogues. These heterocycles exhibit characteristic pH-dependent UV-visible absorption and fluorescence properties that may be utilized for in-situ pH-sensing in biological experiments. The pKa values for this series of compounds, ranging from 4.5 to 7.5, can be systematically varied by manipulating the substitutions on the quinoxaline ring. These novel fluorescent pH indicators are readily available in one synthetic step from commercially available starting materials therefore being significantly more straightforward and less expensive to synthesize than the current standard fluorescent indicators.
Introduction
The ability of dyes such as litmus and phenolphthalein to change their color in response to a pH change has found widespread application in research and industry.1 However, only fluorescent dyes can provide the greater sensitivity required for optical pH measurements inside live cells. The demand for sensitive intracellular pH indicators2 has spurred the search for improved fluorescent dyes that can sense pH changes within physiological ranges. Intracellular pH is generally between ∼6.8 and 7.4 in the cytosol and ∼4.5 and 6.0 in the cell's acidic organelles. Unlike intracellular free Ca2+ concentrations, which can rapidly change by perhaps 100-fold, the pH inside a cell varies by only fractions of a pH unit, and such changes may be quite slow. Although the optical change of even the best fluorescent pH probes is usually relatively small, they have proven to be effective tools for investigating the
role of intracellular pH in diverse physiological and pathological processes, including cell proliferation,3 apoptosis,4,5 fertilization,6 malignancy,7 multidrug resistance,8,9 ion transport,10–12 lysosomal storage disorders and Alzheimer's disease.13 It would be desirable to provide compounds which are effective as pH-dependent fluorescent dye indicators, particularly at low pH. To this end, Diwu and coworkers recently reported14 the development and pH-dependent fluorescence of pyridyloxazole dye (PDMPO) and demonstrated its ability to selectively label acidic organelles. We herein disclose our efforts in this area, on the synthesis, structural characterization and physical properties of a class of heterocyclic
betaines, pyrido[1,2-a]quinoxalines, which exhibit fluorescence which is sensitive to changes in pH in a relevant physiological range.
Results and discussion
Synthesis of pyrido[1,2-a]quinoxalines
The reaction of pyrylium-2-carboxylates with 1,2-phenylenediamines affords the tricyclic 6-oxidopyrido[1,2-a]quinoxalin-11-ium ring system and although the UV spectrum of one analogue has been reported there has been little further study done on this interesting mesoionic heterocycle.15–17 We undertook the synthesis of pyrido[1,2-a]quinoxalines through the reaction of various o-diaminoaromatics 2 with commercially available 4,6-diphenyl-2-ethoxycarbonylpyrylium triflate (1) (Scheme 1). We generally found that conducting the reaction in methanol at room temperature or refluxing acetic acid was optimal, the reaction being complete in a few hours with the triflate salt of the heterocyclic product often crystallising from the reaction medium. 4,5-Disubstituted 1,2-phenylenediamines along with 1,2-phenylenediamine
itself gave single products 3a–d (see Table 1) however 4-monosubstituted diamines 2e, 2g and 2i gave approximately 1 ∶ 1 mixtures of the two possible regioisomers 3e,f, 3g,h and 3i,j which could be readily separated by flash-chromatography to afford the neutral zwitterionic pyrido[1,2-a]quinoxalines. To distinguish between the two isomers a series of NOE experiments were carried out on the trifluoromethyl derivatives 3g and 3h.‡ It was clearly demonstrated that the more polar isomer by silica gel chromatography is the 2-trifluoromethyl isomer 3g and that this relationship held for the chloro 3e,f and nitro 3i,j analogues also. This assignment of isomers was subsequently validated by X-ray crystal analysis (vide infra). Reaction of 3-hydroxy-1,2-phenylenediamine with the pyrylium salt of 1 afforded a single isomer. This was believed to be the 4-hydroxy isomer 3k since the 1-hydroxy isomer 3l would contain a unfavorable steric interaction between the hydroxy group and the 10-phenyl substituent and this was subsequently confirmed
by X-ray analysis of a single crystal of compound 3k.
Table 1 Pyrido[1,2-a]quinoxalines 3
| Compound |
R1 |
R2 |
R3 |
R4 |
|
3a
|
H |
H |
H |
H |
|
3b
|
H |
Me |
Me |
H |
|
3c
|
H |
Cl |
Cl |
H |
|
3d
|
H |
NO2 |
NO2 |
H |
|
3e
|
H |
Cl |
H |
H |
|
3f
|
H |
H |
Cl |
H |
|
3g
|
H |
CF3 |
H |
H |
|
3h
|
H |
H |
CF3 |
H |
|
3i
|
H |
NO2 |
H |
H |
|
3j
|
H |
H |
NO2 |
H |
|
3k
|
H |
H |
H |
OH |
|
3l
|
OH |
H |
H |
H |
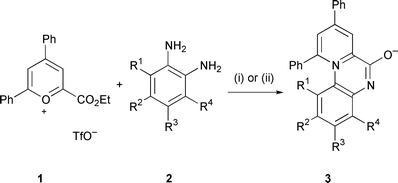 |
| | Scheme 1 (i) AcOH, reflux; (ii) MeOH, room temp. | |
X-Ray crystallography
Several of the pyrido[1,2-a]quinoxalines synthesized during the present study gave single crystals suitable for X-ray diffraction analysis. The crystal structures of 2-trifluoromethyl analogue 3g, 2-nitro analogue 3i and 4-hydroxy analog 3k are represented in Fig. 1. Compounds 3i and 3k were studied as triflate salts. These structures provide conclusive confirmation of the assignment of substituent position as determined previously by NMR experiments. Structural details are similar for the three analogues. Looking at compound 3g in more detail, the geometry of the tricyclic core is significantly distorted from that expected for a fully aromatic system. In particular, the distances around C6,
1.500(6) to C14, 1.342(5) to N5, and about N1, 1.385(5) to C10, 1.432(5) to C12, 1.373 to C14(5) are consistent with significant localization of electron density. The tricyclic pyrido[1,2-a]quinoxalinium system is not planar, but rather consists of two flat sections twisted at the central ring. The dihedral angle between the planes defined by rings A and C is 21.3 (1)°. Phenyl ring E is rotated 53.2(1)° out of the ring C plane while phenyl ring D is rotated 20.8(1)° out of the same plane. These distortions impart a helical shape to the core of the molecule. Fig. 2 illustrates the π-stacking arrangement for 3g. For the lower and middle molecules the separation between the planes defined by atoms N5, C6, O6, C14, N1, and C7 is 3.27 Å, while for the middle and upper molecules it is 3.48 Å. The π-stacking places the oppositely charged atoms O6 and N1 in close proximity. The relevant
distances are 3.360(5) Å between those atoms in the lower pair and 3.714(5) Å in the upper two. Unlike the 3g structure, there is no π-stacking arrangement for the other analogues which are cations in the crystal lattice. The intermolecular interactions for these triflate salts are dominated by hydrogen bonding contacts.
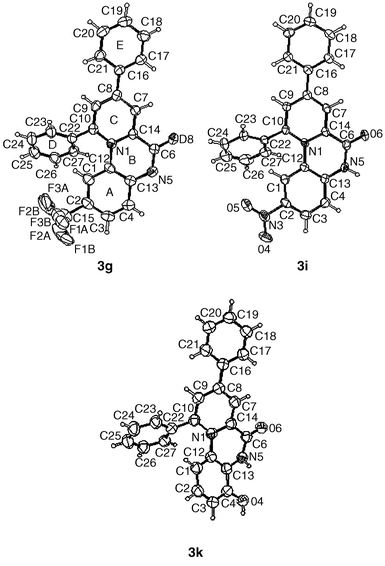 |
| | Fig. 1 Views of 3g, 3i and 3k from their crystal structures showing the numbering scheme employed. Anisotropic displacement ellipsoids for non-hydrogen atoms are shown at the 50% probability level. Hydrogen atoms are displayed with an arbitrarily small radius. | |
 |
| | Fig. 2 A stereo view of molecules of 3g illustrating the π-stacking. The molecule in the middle of the sandwich is at symmetry position x,y,z. The one below is at symmetry position 1 −
x,−y,−z; above symmetry position −x,−y,−z. Hydrogen atoms are omitted for clarity. | |
Membrane permeability study
The permeation of selective pyridoquinoxalines through the lipid membrane was evaluated in the artificial membrane permeability model (see Experimental section for details). The permeability values for the selected compounds, shown in Table 2, fall in the range of medium (>10−6 cm s−1) to high (>10−5 cm s−1). For comparison, the permeability for Rhodamine 123 and calcein acetoxymethyl ester (calcein AM), two fluorescence probes widely used for intra-cellular microscopic imaging studies and cell-based assays,18 was also determined in the artificial membrane system. The compounds of interest here all have a comparable or higher (passive) membrane permeability than that for Rhodamine 123 or calcein AM in aqueous buffer at pH 7. Most likely, these compounds would penetrate cell membrane in the time frame comparable to that for routine cellular experiments
involving the rhodamine or calcein AM dye. It is beyond the scope of this study to address the issues of efflux transporters and compound stability in the biological system, both of which can affect the utilities of these compounds as intracellular pH probes.
Table 2 Membrane permeability, UV absorption and fluorescence emission properties for selective pyridoquinoxalines compounds 3a–k
| Compound |
Permeability/cm s−1 |
λ
max; AC (pH 3.5)b |
λ
max; AC (pH 9.6) |
λ
em;Rel. int. (pH 3.5)c |
|
Nt = not tested.
λ
max
= wavelength at absorption maximum (in nm). AC = molar absorption coefficient (in cm−1 M−1).
λ
em
= wavelength of emission maximum (in nm, excitation at 350 nm). Rel Int = Relative Intensity with respect to compound 3a.
|
|
3a
|
3.9 × 10−6 |
344; 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
319; 33000 |
566; 1.00 |
|
3b
|
2.4 × 10−5 |
333; 13000 |
319; 15000 |
565; 0.47 |
|
3c
|
Nta |
350; 20000 |
322; 25000 |
564; 0.89 |
|
3d
|
1.7 × 10−5 |
375; 20000 |
334; 20000 |
Not detected |
|
3f
|
5.1 × 10−6 |
Nt |
Nt |
Nt |
|
3g
|
2.9 × 10−5 |
353; 21000 |
319; 28000 |
553; 1.02 |
|
3h
|
3.9 × 10−6 |
351; 22000 |
320; 27000 |
554; 0.71 |
|
3i
|
1.5 × 10−5 |
333; 22000 |
334; 26000 |
567; 0.47 |
|
3k
|
1.9 × 10−5 |
343; 14000 |
318; 13000 |
474; 0.20 |
| Rhodamine 123 |
7.7 × 10−6 |
Nt |
Nt |
Nt |
| Calcein AM |
4.0 × 10−6 |
Nt |
Nt |
Nt |
The pyrido[1,2-a]quinoxalines are all intensely colored and show pH-dependent color changes visible to the human eye. Table 2 lists absorption maxima and molar absorption coefficients for compounds 3 under both acidic (pH = 3.5) and basic (pH = 9.6) conditions. Shown in Fig. 3 are a series of UV-visible spectra obtained in systematically varied pH buffers, ranging from 2 to 11, for the parent compound 3a. It is evident from the spectral changes and well-defined isosbestic points that there is only one ionization step in the observed pH range and that both chemical forms are stable. All the compounds in Table 2 exhibit pH-dependent spectral changes, but respond quite differently in terms of the shape of the spectra and the magnitude of the change. The ionization constant can be readily
estimated using the spectrophotometric pKa method of Albert and Serjeant.19 The results are shown in Table 3. The nature of the functional groups on the quinoxaline ring significantly affects the pKa value. For example, the electron-withdrawing nitro function shifted the pKa value from 7.4 for the unsubstituted parent compound 3a to 5.9 for the mono-substituted compound 3i and 4.5 for the di-substituted compound 3d. By manipulating the electronic property of the tricyclic ring, we have synthesized a series of novel pH-sensitive dyes covering a usable pH range from 3 to 9.
Table 3 Aqueous pKa's obtained by the spectrophotometric method of Albert and Serjeant (see ref. 19)
| Compound 3 |
pKa |
|
a
|
7.4 |
|
b
|
7.5 |
|
c
|
6.3 |
|
d
|
4.5 |
|
g
|
6.5 |
|
h
|
6.6 |
|
i
|
5.9 |
|
k
|
6.9 |
|
m
|
5.1 |
Fluorescence studies
Except for compound 3d, all compounds listed in Table 1 are brightly fluorescent in acidic media with diminishing emission intensity and a small red shift toward more basic conditions. Maximum emission wavelengths and fluorescence intensities (relative to compound 3a) for selected compounds 3 are listed in Table 2. Representative fluorescence emission spectra of compound 3a from 450 to 700 nm at an excitation wavelength of 375 nm in pH buffers are shown in Fig. 4. The plots of peak emission intensity as a function of pH (result not shown here) for the compounds in Table 1 are sigmoidal in shape with the inflection point approximately at the pKa for the compound.
 |
| | Fig. 4 Fluorescence emission spectra of compound 3a in (from top to bottom) pH 3.5, 4.5, 5.0, 6.5, 7.0, 7.5, 8.5 and 11.1 buffer. The excitation wavelength was at 375 nm. The intensity is in arbitrary units. | |
Of particular interest is the mono-nitro substituted compound 3i for which selected fluorescence excitation spectra in buffers of different pH values are shown in Fig. 5. The significant red shift from acidic to basic environment allows accurate ratiometric excitation measurement. The plot of intensity ratios at 458 nm/364 nm (purposely chosen to match the wavelengths of commonly available UV and visible lasers) vs. pH's, shown in Fig. 5 (inset), indicates that the ratiometric technique is well-suited for in situ pH measurement in the pH 4–8 range. Compound 3i is a better alternative to existing pH-sensitive ratioable dyes, e.g., DM-NERF and di-Cl-NERF,20 for applications such as confocal fluorescence microscope imaging of intra-cellular pH changes.23
 |
| | Fig. 5 Excitation spectra of compound 3i in (from top to bottom at 364 nm) pH 3.5, 4.5, 5.5, 6.0, 6.5, 7.0 and 8.5 buffers. The fluorescence emission was monitored at 575 nm. The inset shows the fluorescence intensity ratio at the excitation wavelength of 458 nm over 364 nm vs. pH. Note that the intensity ratio at a particular pH is not affected by the total fluorescence. | |
Conclusion
The most attractive feature of the use of pyrido[1,2-a]quinoxalines as pH-dependant fluorescent probes is their ease of synthesis (one single step from commercial reagents) particularly when compared to the multi-step preparation of the PDMPO dye mentioned previously.14 Also of great value is the ability to prepare many, diverse analogs wherein tuning of the electronic properties of the tricyclic ring system is made possible by altering the nature of the attached functional groups. This can vary the pKa values for these pyrido[1,2-a]quinoxalines by at least three log units.
Experimental24
Preparation of pyrido[1,2-a]quinoxalines
See Table 4 for analytical data.
Table 4 Analytical data for pyrido[1,2-a]quinoxalines 3a–k
| Compound (Formula) |
Method |
Yield (%) |
Mp/°C (solvent) |
Found (%) (Required) |
| |
|
|
|
%C |
%H |
%N |
|
Significant darkening up to decomposition temperature.
|
|
3a (C24H16N2O·CHF3O3S·CH3OH) |
A |
88 |
206–208 (MeOH) |
58.57 (58.86) |
3.61 (3.99) |
5.32 (5.28) |
|
3b (C26H20N2O·CHF3O3S·0.75 CH2Cl2) |
A |
79 |
265–266 (MeOH–DCM) |
56.40 (56.47) |
4.11 (3.84) |
5.07 (4.75) |
|
3c (C24H14Cl2N2O·CHF3O3S) |
A |
84 |
283–285 (MeOH) |
52.70 (52.92) |
3.00 (2.66) |
4.72 (4.94) |
|
3d (C24H14N4O5·0.5 CH3OH) |
A |
75 |
274 dec. (MeOH–Et3N) |
64.72 (64.76) |
3.14 (3.55) |
12.25 (12.33) |
|
3e (C24H15ClN2O·CH2Cl2·0.5 CH3OH) |
B |
31 |
162 dec.a (MeOH–DCM) |
63.61 (63.31) |
4.23 (3.96) |
6.19 (5.79) |
|
3f (C24H15ClN2O·CH2Cl2) |
B |
36 |
140 dec.a (MeOH–DCM) |
64.50 (64.19) |
3.83 (3.66) |
6.21 (5.99) |
|
3g (C25H15F3N2O·2H2O) |
B |
27 |
175 dec.a (MeOH–DCM) |
66.61 (66.37) |
3.95 (4.23) |
6.18 (6.19) |
|
3h (C25H15F3N2O·CH2Cl2) |
B |
32 |
180 dec.a (MeOH–DCM) |
62.63 (62.29) |
3.77 (3.42) |
5.77 (5.59) |
|
3i (C24H15N3O3·CHF3O3S·CH3OH·0.5 H2O) |
B |
46 |
222 dec.a (DCM–EtOAc–MeOH) |
53.51 (53.43) |
3.44 (3.62) |
7.18 (7.19) |
|
3j (C24H15N3O3·CHF3O3S·0.5 CH3OH) |
B |
46 |
216 dec.a (DCM–EtOAc–MeOH) |
54.33 (54.74) |
3.46 (3.24) |
7.49 (7.51) |
|
3k (C24H16N2O2·CHF3O3S·CH3OH) |
A |
37 |
272–273 (MeOH) |
57.13 (57.14) |
3.56 (3.87) |
5.48 (5.13) |
Method A: e.g., preparation of 8,10-diphenyl-5,6-dihydro-6-oxopyrido[1,2-a]quinoxalin-11-ium 3a trifluoromethanesulfonate.
To a solution of pyrylium salt 1 (454.4 mg, 1.0 mmol) in anhydrous methanol (8 cm3) was added 1,2-phenylenediamine (119 mg, 1.1 mmol) and the mixture was left standing for 6 days at room temperature. The resulting yellow crystals were filtered and rinsed with dichloromethane to provide pyrido[1,2-a]quinoxaline 3a which was combined with a second crop obtained by dilution of the methanolic mother liquor with an equal volume of dichloromethane followed by filtration (438 mg, 88%) as a yellow solid.
Method B: e.g., preparation of 8,10-diphenyl-6-oxido-3-trifluoromethylpyrido[1,2-a]quinoxalin-11-ium 3h and 8,10-diphenyl-6-oxido-2-trifluoromethylpyrido[1,2-a]quinoxalin-11-ium 3g.
To a solution of pyrylium salt 1 (454.4 mg, 1.0 mmol) in anhydrous methanol (10 cm3) was added 4-trifluoromethyl-1,2-phenylenediamine (193.7 mg, 1.1 mmol) and the resulting mixture was heated at 75 °C (oil-bath temp.) for 1 h. The solvent was evaporated and the residue was purified by chromatography on silica gel with dichloromethane–ethyl acetate–methanol (9 ∶ 9 ∶ 2) to provide 8,10-diphenyl-5,6-dihydro-6-oxo-3-trifluoromethylpyrido[1,2-a]quinoxalin-11-ium 3g (112 mg, 27%) as a red solid. Further elution with dichloromethane–ethyl
acetate–methanol (9 ∶ 9 ∶ 2) afforded 8,10-diphenyl-5,6-dihydro-6-oxo-2-trifluoromethylpyrido[1,2-a]quinoxalin-11-ium 3h (134 mg, 32%) as a red solid.
Crystal structure determination of compound 3g25
Crystals of 3g were recrystallised from dichloromethane–ethyl acetate–hexanes as red needles. A single crystal (0.06 × 0.24 × 0.30 mm) was mounted in inert oil and transferred to the diffractometer.
Crystal data.
C25H15F3N2O, M
= 416.39, monoclinic, a
= 7.045(3), b
= 10.615(4), c
= 25.694(10) Å, β
= 92.68(3)°. U
= 1919.3(13) Å3, T
= 293(2) K, space group P21/n, Z
= 4, μ(Mo–Kα) = 0.917 mm−1, 3418 reflections measured, 2843 unique (Rint
= 0.090) which were used in all calculations. The final residuals were R1 = 0.060 (2813 data; I > 2σI) and wR(F2) = 0.241 (all data).
Crystal structure determination of compound 3i25
Crystals of 3i were recrystallised from dichloromethane–ethyl acetate–methanol as yellow plates. A single crystal (1.20 × 0.50 × 0.20 mm) was mounted in inert oil and transferred to the diffractometer.
Crystal data.
[C24H18N3O3]+ [CF3SO3]−. H2O, M
= 561.48, triclinic, a
= 8.454(2), b
= 9.474(2), c
= 17.001(3) Å, α
= 74.91(3), β
= 85.33(3), γ
= 68.33(3)°. U
= 1221.6(4) Å3, T
= 223(2) K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z
= 2, μ(Mo–Kα) = 0.208 mm−1, 13511 reflections measured, 3303 unique (Rint
= 0.118) which were used in all calculations. The final residuals were R1 = 0.071 (2998 data; I > 2σI) and wR(F2) = 0.185 (all data).
, Z
= 2, μ(Mo–Kα) = 0.208 mm−1, 13511 reflections measured, 3303 unique (Rint
= 0.118) which were used in all calculations. The final residuals were R1 = 0.071 (2998 data; I > 2σI) and wR(F2) = 0.185 (all data).
Crystal structure determination of compound 3k25
Crystals of 3k were recrystallised from dichloromethane–ethyl acetate–methanol as colourless prisms. A single crystal (0.58 × 0.24 × 0.22 mm) was mounted in inert oil and transferred to the diffractometer.
Crystal data.
[C24H17N2O2]+[CHF3SO3]−. CH3OH, M
= 546.51, triclinic, a
= 7.633(3), b
= 10.495(4), c
= 15.251(6) Å, α
= 98.61(3), β
= 90.52(3), γ
= 93.99(3)°. U
= 1204.8(8) Å3, T
= 223(2) K, space group P, Z
= 2, μ(Mo–Kα) = 0.205 mm−1, 9167 reflections measured, 4216 unique (Rint
= 0.0176) which were used in all calculations. The final residuals were R1 = 0.082 (3587 data; I > 2σI) and wR(F2) = 0.205 (all data).
Artificial membrane permeability assay
The artificial membrane permeability assay was developed at GlaxoSmithKline based on the classical microfiltration filter–black lipid membrane technique21 and the contemporary high throughput parallel chemical analysis approach22 with the underlying purpose of developing a high-throughput physico-chemical technique for evaluating drug intestinal absorption and membrane penetration. Conceptually and experimentally, the artificial phospholipid membrane technique is similar to the widely used Caco-2 cell monolayer permeation technique. In short, egg phosphatidyl choline (2%) and cholesterol (1%) are dissolved in n-decane. A small amount of the volatile mixture is applied to the bottom of the microfiltration filter inserts. Phosphate buffer (0.05 M, pH 7.05) is quickly added to the donors and receivers, and the lipids are allowed to form self-assembled lipid bilayers across the small holes in the
filter. The permeation experiment is initiated by spiking the compounds of interest to the donor sides and the the experiment is stopped at a pre-determined elapsed time. The samples are withdrawn and transferred to appropriate vials for analysis by HPLC with UV detection.
References
- H. S. Freeman and J. Sokolowska, Rev. Prog. Color. Relat. Top., 1999, 29, 8 Search PubMed.
- A. W. Czarnick, Chem. Biol., 1995, 2, 423 CrossRef CAS.
- E. A. Schutle, A. Hohendahl, H. Stegmann, J. R. Hirsch, H. Saleh and E. Schlatter, Cell. Physiol. Biochem., 1999, 9, 310 Search PubMed.
- D. Pérez-Sala, D. Collado-Escobar and F. Mollinedo, J. Biol. Chem., 1995, 270, 6235 CrossRef CAS.
- J. Li and A. Eastmann, J. Biol. Chem., 1995, 270, 3203 CrossRef CAS.
- K. Suzuki, Y. Tanaka, Y. Nakajima, K. Hirano, H. Itoh, H. Miyata, T. Hayakawa and K. Kinosita, Jr., Biophys. J., 1995, 68, 739 Search PubMed.
- G. R. Martin and R. K. Jain, Cancer Res., 1994, 54, 5670 Search PubMed.
- S. Simon, D. Roy and M. Schindler, Proc. Natl. Acad. Sci. USA, 1994, 91, 1128 CAS.
- P. D. Poepe, Li Yong Wei, J. Cruz and D. Carlson, Biochemistry, 1993, 32, 11042 CrossRef.
- H. Zhao, X. Xu, J. Diaz and S. Muallem, J. Biol. Chem., 1995, 270, 19599 CrossRef CAS.
- S. A. Levine, S. K. Nath, C. H. C. Yun, J. W. Yip, M. Montrose, M. Donowitz and C. M. Tse, J. Biol. Chem., 1995, 270, 13716 CrossRef CAS.
- R. M. Shepherd and J.-C. Henquin, J. Biol. Chem., 1995, 280, 7915.
- T. A. Davise, R. E. Fine, R. J. Johnson, C. A. Levesque, W. H. Rathbun, K. F. Seetoo, S. J. Smith, G. Strohmeier, L. Volicer, L. Delva and E. R. Simons, Biochem. Biophys. Res. Commun., 1994, 194, 537 CrossRef.
- Z. Diwu, C.-S. Chen, C. Zhang, D. H. Klaubert and R. P. Haugland, Chem. Biol., 1999, 6, 411 CrossRef CAS.
- Y. P. Andreichikov, N. V. Kolovda and G. N. Dorofeenko, Khim. Geterotsikl. Soedin., 1975, 11, 1587 Search PubMed.
- L. N. Chernavskaya, N. V. Kolovda, S. G. Blagorodov and N. A. Dmitrieva, Khim.-Farm. Zh., 1983, 18, 700 Search PubMed.
- P. Molina, M. Alajarin, M. Loranzo-Pena, A. Tarraga and M. J. Vilaplana, J. Heterocycl. Chem., 1984, 21, 1609 Search PubMed.
-
R. P. Haugland, Handbook of Fluorescence Probes and Research Chemicals, Molecular Probes, Eugene, Oregon, 1998 Search PubMed.
-
A. Albert and E. P. Serjeant, The Determination of Ionisation Constants, Chapman and Hall, London, 1984 Search PubMed.
-
R. P. Haugland, Handbook of Fluorescence Probes and Research Chemicals, Molecular Probes, Eugene, Oregon, 1998 Search PubMed.
- M. Thompson, R. B. Lennox and R. A. McClelland, Anal. Chem., 1982, 54, 76 CAS.
- M. Kansy, F. Senner and K. Gubernator, J. Med. Chem., 1998, 41, 1007 CrossRef CAS.
- Both DM-NERF and di-Cl-NERF need to be excited at 488 and 514 nm
for ratiometric imaging. An important logistic issue is that a confocal fluorescence system with an UV and visible laser of the appropriate wavelengths would be more readily available than a system with two Ar lasers (redundant) or 1 Ar laser with dynamic mode switching between 488 and 514 nm (technically challenging). (R. Moosher, personal communication).
- A table of NMR data for compounds 3a–k is given in the supplementary material.
- Crystallographic data for compounds 3g, 3i and 3k are given in the supplementary material. CCDC reference numbers 162091–162093. See http://www.rsc.org/suppdata/p2/b1/b102755g/
for crystallographic files in .cif or other electronic format.
|
| This journal is © The Royal Society of Chemistry 2002 |
Click here to see how this site uses Cookies. View our privacy policy here. 
