Quantitative characterisation of the hydrogen bonding behaviour on the acceptor functions in oligopeptide derivatives with a fluorinated alcohol
Monika Plass* and Ingrid Schaller
Institut für Physikalische Chemie, Martin-Luther-Universität Halle-Wittenberg, Kurt-Mothes-Str. 2, 06120 Halle (Saale), Germany
First published on 22nd November 2001
Abstract
The equilibrium constants for the association of a fluorinated alcohol, 1,1,1,3,3,3-hexafluoropropanol with amino acid and peptide derivatives dissolved in methylene chloride were calculated. The mathematical approach for the determination of the equilibrium constant KOH is based on the decrease of the integral intensity of the OH stretching signal in the infrared spectrum. Alternatively, the decrease of the intensities of the acceptor signals can be used for the calculation of individual equilibrium constants KZ, KPeptide and KEster, for the association of the alcohol on the urethane, peptide and ester function, respectively. The equilibrium constants obtained with both approaches for a number of amino acid, di- and tripeptide derivatives will be discussed. Generally, in no case was the ester function involved in a complex formation with the fluorinated alcohol. For most examples the association constant for the peptide function was larger than for the urethane group. The investigation of the effect of the variation of the concentration on the equilibrium constant shows that up to a ratio cA0/cB0 of the initial concentrations of the alcohol, cA0, and the acceptor compound, cB0, equal to 10, no formation of 1 ∶ 2 or 1 ∶ 3 associates need to be considered.
Introduction
Hydrogen bonding properties of a peptide molecule are not just important for the formation of secondary structures, but also for numerous processes such as octanol–water partitioning, solubilities in water and in organic solvents. Also HPLC retention behaviour is rationalised in terms of hydrogen bond acidity and basicity. Thus, it is not surprising that there have been several attempts to scale hydrogen bond properties either by correlating spectroscopic or thermodynamic data. The first solute hydrogen bond basicity scale based on the 1 ∶ 1 complexation of bases with 4-fluorophenol in carbon tetrachloride was set up by Taft et al.1 In a similar way also the hydrogen bond acidity can be scaled.2,3 Extensive studies on the relationship between hydrogen bond acidity and basicity in the 1 ∶ 1 complexes showed that there exists a linear correlation between the acidity α2H and the basicity β2H of any proton donor and proton acceptor and the logarithm of their equilibrium constant for 1 ∶ 1 complexation measured in carbon tetrachloride as solvent.4–7 This correlation reveals the possibility of approximating equilibrium constants for any 1 ∶ 1 complex of a proton acid and base with known α2H and β2H. Unfortunately, many solutes are not sufficiently soluble in the apolar carbon tetrachloride. Recent attempts were successful in applying an analogous correlation for methylene chloride solutions.8The equilibrium constant for the 1 ∶ 1 complexation of an alcohol and a proton acceptor molecule can be derived from infrared measurements using the Lambert–Beer law and is based on the study of the decrease of the intensity of the OH stretching signal.9 However, when applying this approach problems may arise because of the nature of oligopeptides. In contrast to acceptor compounds generally used for the investigation of equilibrium constants, a protected tripeptide derivative contains at least four C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O acceptor functions with comparable hydrogen bond basicity. Regarding the accessibility of the acceptor functions, differences are expected depending on the bulkiness of the amino acid residues and the location of the acceptor function within the amino acid sequence.
O acceptor functions with comparable hydrogen bond basicity. Regarding the accessibility of the acceptor functions, differences are expected depending on the bulkiness of the amino acid residues and the location of the acceptor function within the amino acid sequence.
Up to now, there have been only a very few theoretical approaches to determining equilibrium constants for polyfunctional bases interacting with an alcohol molecule.10,11 Unfortunately, these methods can not be applied here since the conformational flexibility of the peptide backbone makes the entropy a relevant factor.
Further difficulties arise due to the relatively weak proton acceptor properties of the oligopeptide derivative. Hence, in order to obtain accurate equilibrium constants, strong proton donors, such as fluorinated alcohols, are required which have the disadvantage of influencing the secondary structure of peptides and proteins. Thus, especially at low concentrations, higher ordered arrangements in the peptide backbone were observed due to the helicogenic effect.12,13
Protected di- and tripeptide derivatives exist in an unordered arrangement in solutions of methylene chloride and 1,1,1,3,3,3-hexafluoroisopropyl alcohol (HFiP). However, as the amide I band profiles indicate, the population of the stable conformers is different in the two solvents due to the properties of the solvents.14
In this paper we will show how the equilibrium constants for the 1 ∶ 1 complexation of the selective acceptor functions in oligopeptide and amino acid derivatives with alcohol can be calculated. In order to locate the potential acceptor functions for an alcohol attack we started with a qualitative study using 1H NMR and IR titration.
Theory
The calculation of equilibrium constants for the interaction of a proton donor A and a proton acceptor B from infrared measurements is based on the decrease of the intensity of an association sensitive signal such as the OH stretching vibration. Thus, using the Lambert–Beer law the concentration of the alcohol solution before and after the addition of the acceptor compound can be determined from the integral intensity of the OH signal, eqn. (1) where cass = cA0 − cA, where cA = concentration of the donor compound and cB0 = initial concentration of the acceptor compound. | (1) |
Eqn. (1) is only valid for 1 ∶ 1 complex formation. Self association should be strictly avoided. Furthermore, the interacting donor molecules should only exhibit one active acceptor (donor) function or at least one strongly dominating acceptor function. These conditions limit the applicability of eqn. (1). However, if the acceptor groups can be analysed separately, similar equations to eqn. (1) can be set up studying the intensity decrease of the acceptor carbonyl functions. The sensitivity of these signals will be lower than for the OH signal due to the complex formation with HFiP but nevertheless they represent a ideal tool for the investigation of each acceptor function within a peptide molecule.
In the case of dipeptide derivatives at least three potential acceptor functions a, b and c (urethane CO, peptide CO, ester CO) exist which are similar but not identical in their basicity and accessibility. Thus, at a concentration ratio cAlk0/cPep0 less than 1 the OH group will interact with the acceptor function giving equilibrium constants Ka, Kb and Kc, eqns. (2)–(4).
 | (2) |
 | (3) |
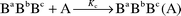 | (4) |
Consequently, for the calculation of the equilibrium constants for each acceptor function, the free alcohol concentration is reduced by the sum of complex concentrations on a, b and c.
Eqn. (1) applied to the acceptor function a can be written as eqn. (5).
 | (5) |
Analogously, Kb and Kc can be calculated. The equilibrium concentration ca is determined from the integral molar absorptivity of preliminarily recorded calibration curves. Our complex model will work based on the following assumptions: (1) The integral absorptivity of the signal does not change upon the addition of HFiP. (2) The concentration of 1 ∶ 2 associates is ignored.
Results and discussion
Qualitative analysis
As stated above, the model for the calculation of the equilibrium constant from the decrease of the acceptor signals of the peptide will only be valid if the amide I signal does not change in its form and position, e.g. the conformation of the peptide backbone does not vary in the presence of the alcohol. However, fluorinated alcohols in particular are known to cause major changes in the peptide backbone even at low concentrations. Previous investigations on the solvent effect on the conformational behaviour of oligopeptides have shown that, due to the ability of HFiP to act as a strong proton donor, while exhibiting almost no proton acceptor properties, the conformational behaviour of the peptide function in an HFiP solution differs significantly from that in solution in water or methylene chloride.14Hence, the first step of our investigation will be focused on the influence of HFiP on the conformational equilibrium of the peptide molecules dissolved in apolar solvents. As a consequence of the proton attack on the carbonyl functions the nearest environment in the peptide backbone will be changed and consequently result in a shift of the 1H NMR signals of the NH and CαH next to the acceptor function. In constrained conformers the relative shift of the NH band can be used for the semi-quantitative description of the equilibrium constant for the hydrogen bonded association complex.151H–1H COSY, 1H–13C COSY and systematic variation of the amino acid composition were used to assign the signals in the 1H NMR spectra (Table 1).
| Substance | CH2OC(O) | NH | CαH | CβH | OCH3 |
|---|---|---|---|---|---|
| a nd = not detectable; the signals are strongly overlapped and cannot be assigned to the different amino acid residues. | |||||
| Z-1Ala-2Ala-OMe | 5.142 | 3.730 | |||
| 1Ala | 5.260 | 4.223 | 1.374 | ||
| 2Ala | 6.429 | 4.561 | 1.374 | ||
| Z-1Ala-2Ala-3Ala-OMe | 5.142 | 3.727 | |||
| 1Ala | 5.265 | 4.215 | |||
| 2Ala | 6.594 | 4.447 | |||
| 3Ala | 6.554 | 4.523 | |||
| Z-1Ala-2Ala-3Val-OMe | 5.143 | 3.718 | |||
| 1Ala | 5.259 | 4.221 | 1.365 | ||
| 2Ala | 6.546 | 4.467 | 1.365 | ||
| 3Val | 6.509 | 4.479 | 2.152 | ||
| Z-1Ala-2Val-3Val-OMe | 5.140 | 3.714 | |||
| 1Ala | 5.308 | 4.241 | 1.359 | ||
| 2Val | 6.567 | 4.241 | |||
| 3Val | 6.377 | 4.501 | |||
| Z-1Val-2Val-3Val-OMe | 5.087 | nda | 3.710 | ||
| 1Val | 5.396 | 4.002 | |||
| 2Val | 6.4545 | 4.261 | |||
| 3Val | 6.4545 | 4.506 | |||
| Z-1Val-2D-Val-3Val-OMe | 5.079 | 3.681 | |||
| 1Val | 5.3435 | 3.9835 | nd | ||
| 2D-Val | 6.474 | 4.3395 | |||
| 3Val | 6.515 | 4.488 | |||
Comparing the 1H NMR data in Table 1 the NH protons of the urethane functions are always found at higher fields than the NH signals of the peptide bond. Furthermore, the chemical shifts of the peptide protons are determined by the type of the amino acid, its position in the peptide backbone and its chirality. For substances with no defined secondary structure the proton chemical shift of NH and CαH will represent the average distribution of the variety of conformers.
The addition of alcohol to an apolar peptide solution will result in a shift of the amide protons. In Fig. 1 the relative chemical shifts of the NH and CαH signals of Z-1Ala-2Ala-OMe are shown vs. the volume of HFiP added to 1 ml of the peptide solution.
 | ||
| Fig. 1 Relative chemical shift changes (Δδ/δ) of 1H NMR signals of Z-1Ala-2Ala-OMe with the addition of HFiP (NH: 1Ala: ▲, 2Ala: ×, CαH: 1Ala: ●, 2Ala: ■). | ||
In cyclopeptides the observed shifts are purely an effect of hydrogen bonding. In linear oligopeptides the observations of the 1H NMR titration must be discussed in a more complex manner. The formation of hydrogen bonds will generally lead to a downfield shift of the NH protons, whereas conformational effects can result in a shift in either field direction. From the comparison of the responses of the NH signals to the attack of HFiP it is possible to derive conclusions on the accessibility of the amide groups within the molecule assuming similar hydrogen bond basicities of the acceptors.
When only 10 μl HFiP were added to 1 ml Z-1Ala-2Ala-OMe solution the urethane signal shifts to lower magnetic field than that found in the pure CD2Cl2 solution. Only by increasing the HFiP concentration does the 1NH signal behave similarly to the 2NH signal and shift to higher magnetic field. Also the CαH signals are affected by the addition of HFiP and are generally shifted to higher field.
As known from the behaviour of cyclopeptides with defined conformational structure the addition of a proton donor will lead to downfield shifts of the NH signals due to the association with the peptide CO functions. However, it needs to be kept in mind that also the conformation of the peptide backbone may be changed by the addition of HFiP. Comparing the overall Δδ/δ the 2NH is shifted more strongly than the urethane NH signal which indicates a preference for intermolecular hydrogen bonding on the peptide group.
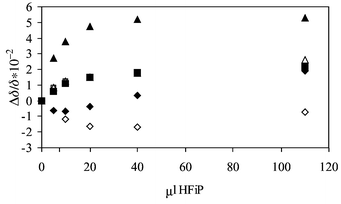
In a methylene chloride solution of the tripeptide derivative Z-1Ala-2Ala-3Ala-OMe the most significant effect was observed for the NH signal in the peptide bond 2Ala-3Ala (Fig. 2). The response of the 2NH is rather low whereas the 1NH (urethane) group shows a shift to higher fields at 10μl HFiP per ml peptide solution, indicating that this function becomes more shielded at low alcohol concentrations. Only at high alcohol amounts (110 μl) is the sign of Δδ/δ positive as expected for a hydrogen bonded species.
 | ||
| Fig. 2 Relative chemical shift changes (Δδ/δ) of the NH protons of Z-1Ala-2Ala-3Ala-OMe with the addition of HFiP (1Ala: ◆, 2Ala: ■, 3Ala: ▲). | ||
Summarising the results of the NMR titration it was found that in di- and tripeptide derivatives the urethane and the peptide functions respond differently to the addition of HFiP. If the relative low field shift of the NH signal can be taken as an indicator of the hydrogen bonding tendency of an individual CO function the following order can be given: 2Ala-3Ala > 1Ala-2Ala > Z-1Ala. It cannot be excluded that conformational changes in the peptide backbone may overlap the effect of hydrogen bond formation and hence contribute to the relative shift of the NH signal. An indication of the occurrence of conformational effects may be seen in the high field shift of the urethane NH with 10μl in solution.
In Fig. 3 the analogous alcohol titration was monitored in the amide I region of the mid infrared region, which is more sensitive to conformational effects in linear oligopeptides than NMR spectroscopy. After application of band deconvolution and peak fitting procedures additional signals at 1710 and 1651 cm−1 were found which could be assigned to the associated species of the urethane and peptide function. It should be mentioned here that at low alcohol concentrations neither does the ester carbonyl function change in its intensity nor could a new signal be observed which would indicate hydrogen bond formation. Considering steric relations we cannot understand this behaviour but maybe by its lower basicity the ester function becomes a non-attractive acceptor group.
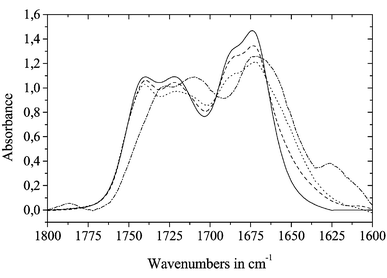 | ||
| Fig. 3 Carbonyl and amide I region of Z-Ala-Ala-Ala-OMe dissolved in methylene chloride and on addition of defined amounts of HFiP (solid line: pure peptide solution, dashed: 1 ml solution + ca. 1μl HFiP, dotted: 1 ml solution + 5 μl HFiP, dash-dotted: 1 ml solution + 110 μl HFiP) | ||
With an amount of 110 μl HFiP per 1 ml solution the spectrum almost exhibits the contours of the spectrum recorded in a solution of pure HFiP (not shown in Fig. 3). Hence the solvent–solute interactions are clearly determined by the alcohol and not by the apolar methylene chloride.
Generally similar behaviour in the 1H NMR titration as discussed for Z-1Ala-2Ala-3Ala-OMe is observed for the diastereoisomers of Z-1Val-2Val-3Val-OMe. However, the accessibility of the amide functions on 2Val and 3Val will be more obstructed than in Z-Ala-Ala-Ala-OMe due to the bulky side chains on Cα. With regard to the accessibility of the peptide functions in LLL and LDL diastereoisomers the urethane and the peptide bond between 1Val and 2Val behave comparably (Fig. 4). Differences were found for the NH function of the 2Val-3Val peptide bond where the LDL diastereoisomer shifts slightly more strongly towards the high magnetic field than the LLL form.
 | ||
| Fig. 4 Relative chemical shift changes (Δδ/δ) of the NH protons of Z-1Val-2Val-3Val-OMe (open symbols) and Z-1Val-D–2Val-3Val-OMe (solid symbols) with the addition of HFiP (1Val: diamond, 2Val: square, 3Val: triangle). | ||
From these experiments it was concluded that, for the determination of the equilibrium constant for HFiP-peptide association by means of infrared spectroscopy, the concentration of HFiP should be small in order to avoid conformational changes in the peptide backbone. As the NMR spectra show the conditions are fulfilled at alcohol concentrations of 0.005 to 0.01 mol l−1.
Band deconvolution and band fitting approaches give us the positions of the acceptor functions in the amino acid and peptide derivatives and the associate bands (Table 2).
| Substance | νEster | νAc/Z | νAc/Zass | νPept. | νPept2 | νPept2ass |
|---|---|---|---|---|---|---|
| Ac-Ala-OMe | 1742.4 | 1678.9 | 1655.7 | |||
| Ac-Phe-OMe | 1743.1 | 1679.6 | 1656.9 | |||
| Ac-Val-OMe | 1739.0 | 1680.6 | 1657.6 | |||
| Z-Ala-Ala-OMe | 1742.3 | 1720.4 | 1705.5 | 1683.9 | 1666.0 | |
| Z-Phe-Phe-OMe | 1743.9 | 1722.8 | 1701.9 | 1682.7 | 1660.0 | |
| Z-Val-Val-OMe | 1739.7 | 1721.2 | 1703.4 | 1682.7 | 1662.7 | |
| Z-Ala-Val-OMe | 1740.2 | 1721.8 | 1705.7 | 1685.8 | 1669.6 | |
| Z-Ala-Ala-Ala-OMe | 1742.8 | 1720.0 | 1707.0 | 1689.4 | 1671.2 | 1649.7 |
| Z-Phe-Phe-Phe-OMe | 1744.4 | 1722.1 | 1701.7 | 1685.7 | 1669.2 | 1652.0 |
| Z-Ala-Val-Val-OMe | 1740.6 | 1720.8 | 1703.2 | 1689.4 | 1671.8 | 1649.5 |
| Z-Val-Val-Val-OMe | 1740.3 | 1721.1 | 1707.1 | 1690.5 | 1672.8 | 1656.4 |
Quantitative analysis
The difficulty in the calculation lies in the occurrence of a number of acceptor functions which are similar in their hydrogen bond basicity. This, however, also implies a challenge for the calculation of individual K values since the acceptor functions show separate signals in the carbonyl stretching region as discussed in the theoretical part.If the concentration ratio cA0/cB0, where cA0 and cB0 are the initial concentration of the alcohol and the peptide, respectively, becomes larger than 1 the chances to form higher complexes and species of the sort Ba(A)Bb(A)Bc, Ba(A)BbBc(A), BaBb(A)Bc(A) and Ba(A)Bb(A)Bc(A) are increased. In comparison with other acceptor molecules peptide derivatives are rather weak acceptor molecules. Hence from the statistical point of view the population of 1 ∶ 1 associated species will be always low for the peptides under the given experimental conditions. Fig. 5 shows K as a function of cA0/cB0.
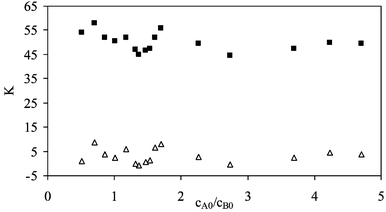 | ||
| Fig. 5 Equilibrium constant Kvs.cA0/cB0 for the proton acceptor functions in Ac-Phe-OMe dissolved in methylene chloride at 25 °C (■: KAc, △: KEster). | ||
Two major conclusions can be drawn from the plot of the equilibrium constant vs.cA0/cB0.
Firstly, the equilibrium constant is constant over a broad range of cA0/cB0. Hence no 1 ∶ 2 association occurs in excess concentration of HFiP.
Secondly, to our surprise the ester function does not show any sign of a complex formation although it should be easily accessible for the proton donor. The equilibrium constant calculated for the association on this position is ca. zero.
The conclusions given above for the amino acid derivatives were confirmed by the plot of Kvs.cA0/cB0 for the association of HFiP with dipeptide and tripeptide derivatives as shown for the example of Z-Ala-Val-OMe in Fig. 6. Again, the equilibrium constant does not change with increasing alcohol concentration and 1 ∶ 2 association can be neglected in the discussion. However, association occurs on different acceptor positions e.g. the urethane and the peptide functions which exist in an equilibrium state. An interaction with the ester function could again not be found and this legitimates the use of the ester carbonyl signal as an internal standard. Attempts to adjust concentration differences by correlation with the NH intensity failed because the extinction coefficient of the NH is obviously influenced by the hydrogen bond formation on the neighbouring CO group. The standard deviation of the equilibrium constants given in Tables 3 to 5 can be given as less than 10%.
 | ||
| Fig. 6 Equilibrium constant Kvs. cA0/cB0 for the proton acceptor functions in Z-Ala-Val-OMe dissolved in methylene chloride at 25 °C (◇: KPeptide, ○: KZ, △: KEster). | ||
Based on the non-occurrence of higher associates the average equilibrium constant for the association on each acceptor site can be given in Table 3 for the amino acid derivatives.
| System | KOH | KEster | KAc/Z |
|---|---|---|---|
| With HFiP | |||
| Ac-Ala-OMe | 86 | 0 | 49 |
| Ac-Phe-OMe | 84 | 0 | 46 |
| Ac-Val-OMe | 80 | 0 | 49 |
| With phenol | |||
| Ac-Val-OMe | 17 | 0 | 11 |
Comparing the equilibrium constants calculated from the decrease of the OH signal which gave KOH and the K value calculated from the change of the intensity of the acceptor signal KAc, large discrepancies become obvious. KOH is almost twice as large as KAc. One explanation might be that besides association on these positions the electron lone pair of the nitrogen atom gets attacked. Also the electron π system can act as an acceptor function.
A further result concerns the peculiarities of the amino acid derivatives. Generally KAc and KOH are similar in magnitude for all acetyl amino acid methyl esters. The small differences are rather caused by the changed basicity than by steric hindrance.
In the case of the dipeptides we observe an association on the urethane and the peptide function giving the equilibrium constants in Table 4.
| System | KOH | KEster | KZ | KPeptide |
|---|---|---|---|---|
| With HFiP | ||||
| Z-Ala-Ala-OMe | 98 | 0 | 6 | 4 |
| Z-Phe-Phe-OMe | 81 | 0 | 5 | 7 |
| Z-Val-Val-OMe | 98 | 0 | 6 | 9 |
| Z-Ala-Val-OMe | 76 | 0 | 7 | 14 |
| With phenol | ||||
| Z-Val-Val-OMe | 11 | 0 | 0 | 1 |
Comparing the KOH data in Table 4 with those in Table 3 no significant changes were found. The different acceptor sites in the dipeptide compete for a complex formation with HFiP. Hence it can be understood why the equilibrium constants KZ and KPeptide are significantly lower than KAc. The magnitude of the equilibrium constant should be determined by two effects: the hydrogen bond basicity and the accessibility of the acceptor function. The relative shift of the acceptor signal due to the association can be taken as an indicator of the hydrogen bond basicity. For Z-Ala-Val-OMe the Δν(COfree − COass) values for the peptide function and the urethane function are 16.2 and 16.1 cm−1, respectively, and confirm that the strength of the hydrogen bonds on the urethane and the peptide function are comparable. Generally, it would have been expected that the acceptor functions in the peptide backbone should be less accessible than functions on the C- and N-terminal ends of the peptide. However, that does not mean that the equilibrium constant of a urethane function has to be larger than that of a peptide function. In fact, it is only observed that KZ > KPeptide for Z-Ala-Ala-OMe. The other dipeptide derivatives show the opposite effect: KZ < KPeptide.
With enlarging the peptide chain by another amino acid residue the calculation of the individual equilibrium constants based on the decrease of the band intensity of the acceptor functions becomes complicated because of the presence of conformational equilibria in the peptide bond. Thus, negative equilibrium constants for the association with Z-Val-Val-Val-OMe with phenol and HFiP are calculated from the intensity change of the signal at 1689.7 cm−1 (Table 5). Obviously the complexation of one acceptor position in the molecule does cause a change in the peptide backbone which results in an increase of the extinction coefficient of the signal at 1689.7 cm−1 and makes it impossible to obtain reliable equilibrium constants for this molecule. Note that this happens independently on the proton donor used in the study only for Z-Val-Val-Val-OMe which is a peptide with a branched side chain on Cβ. The 1H NMR titration indicated that the change in the conformation of the peptide backbone as a consequence of the hydrogen bond formation might be possible. The occurrence of negative equilibrium constants demonstrates the limitations of the approach in calculating the equilibrium constants from the decrease of the acceptor signal.
| System | KOH | KEster | KAc/Z | KPeptide 1 | KPeptide 2 |
|---|---|---|---|---|---|
| With HFiP | |||||
| Z-Ala-Ala-Ala-OMe | 0 | 9 | 6 | 13 | |
| Z-Phe-Phe-Phe-OMe | 132 | 0 | 6 | 6 | 16 |
| Z-Val-Val-Val-OMe | 147 | 0 | 6 | (−8) | 21 |
| Z-Ala-Val-Val-OMe | 216 | 0 | 5 | 8 | 12 |
| With phenol | |||||
| Z-Val-Val-Val-OMe | 14 | 0 | 1 | (−7) | 7 |
The tendency of favoured hydrogen bond formation is obvious: the preferred acceptor sites are the peptide functions. This occurs also if the acceptor groups are obstructed due to bulky side chains. Thus, the conformational equilibrium will be changed. For the ester function we have no hint that any hydrogen bonding takes place.
The question now arises of what to expect when the peptide chain is further extended by one amino acid residue. In those cases the formation of the first loop of a helical structure can be realised in formation of a ten membered ring, C10. The energetic stabilisation of the molecule is, however, no more effective than the C7 rings.16 In methylene chloride C7 rings were not observed and for those reasons C10 rings should not occur either. As long as the attack of a proton donor on the preferred peptide acceptor functions is not hindered by the shielding of the apolar side chain residues, the intramolecular hydrogen bonding will be inferior to the intermolecular interaction. Unfortunately, the enlargement of the peptide backbone does drastically lower the solubility of the peptide in apolar solvents.
Experimental
Materials
The acetyl amino acid, dipeptide and tripeptide derivatives were purchased from Bachem Biochemica (Heidelberg). Their enantiomeric purity exceeded 99%.The benzyloxycarbonyl protected homodipeptide and homotripeptide methyl esters of L-alanine, L-valine and L-phenylalanine were synthesised by C. Griehl, Anhalt University of Applied Sciences, Köthen, as described elsewhere.17,18 The purity of the compounds was checked chromatographically. The diastereoisomeric yield exceeded 95%. After drying all substances were stored in a desiccator to avoid water traces. The solvent methylene chloride (Fluka, HPLC grade) was carefully dried with molecular sieves in order to remove any traces of water. As proton donors 1,1,1,3,3,3-hexafluoroisopropanol (HFiP, Merck, p.a.) and phenol (Fluka, p.a.) were used.
FT-IR measurements
The measurements were performed on an FT-IR spectrometer IFS 25 (Bruker) using a resolution of 2 cm−1. 32 scans were accumulated. For the measurements a 3 mm NaCl cell was placed in a thermostated cell holder at constant temperature of 25 °C. In a first step the pure alcohol dissolved in methylene chloride was recorded. Then, various amounts of the acceptor compound were dissolved in the alcohol solution. Afterwards, the peptide solutions were recorded. The concentration of the alcohol solution was varied in the range of 5 × 10−3 to 10−2 mol l−1. At constant alcohol concentration the amount of the added proton acceptor compound was varied in the range of 1 × 10−3 to 5 × 10−2 mol l−1.Evaluation
The determination of the alcohol and peptide concentration is based on calibration curves which contain the data of at least 20 different measurements of the pure compounds in the concentration range given above. As a measure of the alcohol concentration the integral intensity of the OH signal (3631 to 3478 cm−1) was used. For the determination of the individual acceptor concentration the band profile between 1800 and 1625 cm−1 was analysed by the band fitting procedure which is implemented in the OPUS Vs. 2.0 software package. The successful application of this method requires the determination of the number and position of the individual signals which can result from the band deconvolution using Lorentz functions. More detailed information about the difficulties in the application of this procedure are given in references 19–22. Using the information obtained from the band deconvolution technique an initial set of Gaussian–Lorentzian functions was generated for the fit of the band profile (Fig. 7). The fitting results were accepted when the correlation coefficient was larger than 0.995. The integral intensities of the separated bands were plotted vs. the peptide concentration to give a calibration curve (r2 = 0.995). The band profiles of the alcohol–peptide solutions were fitted using the band width, the type of the Gaussian–Lorentzian profile and the band position as fixed parameters recognising the presence of two complex signals. The equilibrium constants were calculated according to eqn. (2) for the association on the acceptor functions. The constants given in Tables 3–5 are averaged over at least 20 different measurements having a standard deviation of less than 10%. | ||
| Fig. 7 Set of six separate signals used to fit the amide I band profile of Z-Ala-Val-Val-OMe. | ||
1H NMR measurements
1H NMR spectra were recorded on a 400 MHz NMR spectrometer (Varian). Generally 32 scans were accumulated. The HFiP titration was performed as follows: solution of the peptide derivatives in CD2Cl2 at concentrations of 0.008 to 0.01 mol l−1 were prepared. 1 ml of this solution was used to fill a 5 mm NMR tube. The spectrum of this solution was recorded at a temperature of 27 °C. Then, defined volumes of HFiP were added to the solution in the NMR tube and the carefully mixed solution was recorded again. The experiment was repeated until the total amount of 110 μl of HFiP were added to the peptide solution. The relative shifts of the NH and the CαH were used to monitor the association.Acknowledgements
We thank Professor A. Kolbe, Martin-Luther-University, for critical discussion and for reviewing the manuscript. Furthermore, we are indebted to Professor C. Griehl, Anhalt University of Applied Sciences, for donating the peptide derivatives used in this study.This work was supported by the Deutsche Akademie der Naturforscher Leopoldina with funds of the BMTF.
References
- R. W. Taft, D. Gurka, L. Joris, P. v. R. Schleyer and J. W. Rakshys, J. Am. Chem. Soc., 1969, 91, 4801 CrossRef CAS.
- P. L. Sadek, P. W. Carr, R. M. Doherty, M. J. Kamlet, R. W. Taft and M. H. Abraham, Anal. Chem., 1985, 57, 2971 CrossRef CAS.
- M. J. Kamlet, D. J. Abraham, R. M. Doherty, R. W. Taft and M. H. Abraham, J. Pharm. Sci., 1986, 75, 378.
- M. H. Abraham, P. L. Grellier, D. V. Prior, J. J. Morris, P. J. Taylor, C. Laurence and M. Berthelot, Tetrahedron Lett., 1989, 30, 2571 CrossRef CAS.
- M. H. Abraham, P. L. Grellier, D. V. Prior, P. P. Duce, J. J. Morris and P. J. Taylor, J. Chem. Soc., Perkin Trans. 2, 1989, 699 RSC.
- M. H. Abraham, P. L. Grellier, D. V. Prior, J. J. Morris, P. J. Taylor and R. M. Doherty, J. Org. Chem., 1990, 55, 884.
- M. H. Abraham, P. L. Grellier, D. V. Prior, J. J. Morris, P. J. Taylor and R. M. Doherty, J. Org. Chem., 1990, 55, 2227 CrossRef CAS.
- M. Plass, M. H. Abraham and M. Berthelot, unpublished work.
- A. Kolbe and H. Pracejus, Ber. Bunsenges. Phys. Chem., 1966, 70, 883 Search PubMed.
- D. Clotman, D. Van Lerberghe and Th. Zeegers-Hyuskens, Spectrochim. Acta, Part A, 1970, 26, 1621 CrossRef CAS.
- J. P. Muller, G. Vercruysse and Th. Zeegers-Hyuskens, J. Chem. Phys. Phys. Chim., 1972, 1439 Search PubMed.
- O. Pieroni, A. Fissi, C. Pratesi, P. A. Temussi and F. Ciardelli, Biopolymers, 1993, 33, 1 CrossRef CAS.
- N. Hirota, K. Mizuno and Y. Goto, Protein Sci., 1997, 6, 416 CAS.
- M. Plass, C. Griehl and A. Kolbe, J. Mol. Struct., 2001, 570, 203 CrossRef CAS.
- A. Perczel, I. Lengyel, H. H. Mantsch and G. D. Fasman, J. Mol. Struct., 1993, 297, 115 CrossRef CAS.
- S. Krimm, Biopolymers, 1983, 22, 217 CrossRef CAS.
- C. Griehl and S. Merkel, Int. J. Peptide Protein Res., 1995, 45, 217 Search PubMed.
- W. König and R. Geiger, Chem. Ber., 1970, 103, 2024 Search PubMed.
- J. K. Kauppinen, D. J. Moffatt, H. H. Mantsch and D. G. Cameron, Appl. Spectrosc., 1981, 35, 271 Search PubMed.
- J. K. Kauppinen, D. J. Moffatt, H. H. Mantsch and D. G. Cameron, Anal. Chem., 1981, 53, 1454 CrossRef CAS.
- J. K. Kauppinen, D. J. Moffatt, D. G. Cameron and H. H. Mantsch, Appl. Optics, 1981, 10, 1866 Search PubMed.
- M. A. Czarnecki and Y. Ozaki, Spectrochim. Acta, 1996, 52, 1593 Search PubMed.
| This journal is © The Royal Society of Chemistry 2002 |