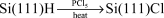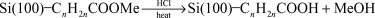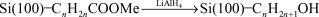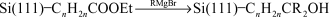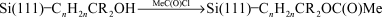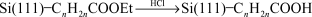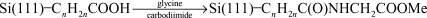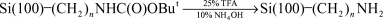Organic modification of hydrogen terminated silicon surfaces1
Danial D. M. Wayner* and Robert A. Wolkow Steacie Institute for Molecular Sciences, National Research Council of Canada, Ottawa, ON K1A 0R6, Canada
Received (in Cambridge, UK) 13th August 2001
First published on 28th November 2001
Abstract
Covering: 1990–2001.
1 Introduction
Over the last decade there has been increasing awareness of the opportunities presented by the convergence of surface science and organic chemistry for the development of revolutionary concepts for the design of molecular scale devices and for the integration of solid state, inorganic structures with biologically active interfaces. Although the reactions of organic molecules with silicon surfaces are not new, this area which traditionally has been dominated by physical scientists has increasingly been influenced by organic and bioorganic chemists. This organic perspective has brought new levels of complexity of structure and function and greater understanding of the molecular basis of reactivity.
This review focuses on the surface chemistry of hydrogen terminated silicon surfaces. Much of the literature related to the surface chemistry of silicon is actually carried out on silicon with its intact native oxide or on “clean” surfaces in ultrahigh vacuum (UHV) chambers. The chemistry of the native oxide and other glasses is well developed and outside of the scope of this review. For many potential molecular electronic applications the oxide is not desirable as it presents an additional insulating barrier between the organic layer and the bulk silicon. In addition, unless grown under carefully controlled conditions, the silicon–silicon oxide interface has a high density of electronic defects which limit its usefulness in future devices. Clean surfaces tend to undergo complex reconstructions associated with the loss of coordination of the silicon atoms at the silicon–vacuum interface. The influence of the electronic and crystallographic structure of the reconstructed
surfaces on the reactivity and selectivity of organics is a subject of ongoing interest and also is beyond the scope of this review. For an overview of organic chemistry on clean silicon surfaces we refer the reader to two reviews which recently have appeared.2,3
The chemical and physical properties of hydrogen terminated silicon surfaces have received more recent attention for a number of reasons. Firstly, the surfaces are reasonably stable and can be prepared and manipulated in air as well as in a number of organic solvents. Thus, high quality materials are available without the need for expensive vacuum systems. Secondly, as you will see below, the chemistry can be understood in many cases by analogy with the chemistry of organosilane molecules for which a vast literature exists. This allows not only an element of rational design of interfaces, but presents opportunities for systematic physical organic studies of the kinetics and mechanisms of reactions. Finally, potential applications in hybrid molecular-semiconductor devices or chemical-biosensors requires a detailed understanding of the interface between silicon and organic materials. This review will cover aspects of the preparation of hydrogen terminated silicon surfaces, their reactions,
and the mechanisms of reactions, to the extent that they are understood. We have tried, where possible, to generalize ideas and concepts and, occasionally to speculate about factors which, in our view require more detailed investigation. It is hoped that this review will stimulate physical organic chemists to think about the opportunities and challenges in this burgeoning area of surface chemistry.
2 Preparation of silicon surfaces
The quality of the starting material for reactions on silicon surfaces is of critical importance. In addition, the structure and electronic properties of the surface must be well understood in order to draw sensible mechanistic conclusions regarding the reactions of organic molecules. Depending on the specific surface, the hydrogen termination is carried out either by reaction of a clean surface with hydrogen atoms in UHV or by chemical etching in aqueous flouride. For simplicity, this review will be restricted to descriptions of two particular silicon surfaces (on which essentially all of the studies in the literature have been carried out). The first is the Si(111) surface for which simple chemical etching procedures are available. The second is the Si(100) surface which is, in fact, the surface upon which commercial microelectronic devices are fabricated. However, the preparation of high quality Si(100) surfaces requires UHV methods.
2.1 Si(111)–H surfaces
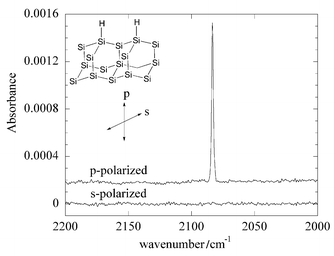
In 1990 Chabal and his coworkers described a simple wet chemical method for the preparation of atomically flat hydrogen terminated Si(111) (henceforth referred to as Si(111)–H).4 They found that the HF etching of Si(111) at pH 8–9 (i.e. ammonium fluoride) resulted in the formation of Si(111)–H in which the Si–H bond is oriented normal to the surface. The observation of a single narrow line at 2083.7 cm−1 (the Si–H stretch) with p-polarized infrared light (i.e. the electric field component is perpendicular to the surface) and the absence of this vibration with s-polarized infrared light (i.e. the electric field component is in the plane of the surface) provided unambiguous proof of the orientation of the Si–H bond (Fig. 1). The procedure was later modified by Wade and Chidsey5 who showed that dissolved oxygen initiated
pitting of the surface since the open circuit potential (i.e. the effective oxidation potential) of the Si(111) surface in ammonium fluoride was sufficiently low to generate superoxide, O2−˙. The superoxide ion then reacts with an Si(111)–H bond on a terrace to initiate the formation of an etch pit. The procedure was further modified by Allongue et al. who showed that surface pitting could be further suppressed by using one-side polished silicon wafers or by abrading part of the polished surface.6 They suggested that the rough surface provides cathodic protection by acting as a sacrificial anode.
Fig. 1 Internal reflection spectra of Si(111)–H in p- and s-polarization.
The surfaces prepared in this way are reasonably stable and atomically flat on the tens of microns scale and are of very high quality, both structurally and electronically. Fig. 2 shows an STM image of a chemically etched Si(111)–H surface. In Fig. 2a two adjacent terraces are separated in height by about 3 Å. Fig. 2b is an atomically resolved image of the same surface showing the three-fold symmetric arrangement of silicon atoms, separated by 3.84 Å, each capped with a hydrogen atom.
Fig. 2 UHV STM images of Si(111)–H prepared by chemical etching in ammonium fluoride. (a) 150 × 150 nm2 image showing terraces and steps. (b) 10 × 10 nm2 image with resolution of silicon atoms.
The mechanism of the ammonium fluoride etching of Si(111) has been the subject of detailed investigation. Chabal and coworkers4,7 and later Behm and his coworkers8 noted that etching with HF at low pH led to rougher surfaces than etching at high pH (i.e. ammonium fluoride). This work led to the widely accepted step-flow mechanism which accounts for the smoothing of the Si(111)–H surface. This mechanism, which has been refined by Allongue et al.6,9,10 and by Hines and coworkers,11–13 is initiated by a rate limiting oxidative addition of hydroxide on a silicon atom at a step edge followed by displacement of the hydroxide by fluoride ion. This leads, eventually to the removal of silicon from the surface (etching) in the form of SiF3OH and the capping of the surface silicon atoms by hydrogen.
The eventual smoothing of the surface leading to well defined steps and terraces (Fig. 2) is a consequence of the relative rates of reaction of hydroxide with different surface sites (Fig. 3). The relevant reactions are shown in eqns. (1) and (2). Using Monte Carlo simulations, Allongue and Kasparian9 and later Flidr and coworkers14 showed that the reaction rates are site specific with kink sites being the most reactive and terrace sites being the least reactive (Table 1). From the model in Fig. 3 it can be seen that the removal of a silicon atom from a kink creates another kink site at the adjacent silicon. Thus, the etching process runs along the step edge resulting in the so-called step-flow mechanism. The preferential etching of kink
sites and the unreactive nature of terrace sites leads to the flattening of the surface at equilibrium. The flattening occurs when a kink is permanently removed when step-flow propagates on the same step in opposite directions and transforms the step from a high reactivity to a low reactivity state. Using a deuterium labeled surface, Luo and Chidsey15 determined the rate of step-flow etching to be 4.2 nm s−1 in Ar sparged ammonium fluoride at 25 °C, or about 10 rows of silicon atoms per second. The rate was measured by the zero order disappearance of the Si–D stretch using FTIR and the average terrace width which was measured by STM.
(1)
(Si)3Si–OH + 3HF → 3(Si)3SiH + SiF3OH
(2)
Fig. 3 (a) Model of the Si(111)H surface showing the vertical monohydride bonds on the terraces and the monohydride step in the (11) direction and a vertical dihydride in the (2) direction. (b) Top view of the Si(111) surface showing the etching sequence during step-flow etching. Removal of silicon atoms from a kink site leads to the formation of a new kink site. Step-flow in opposite directions along the same step leads to the permanent removal of the kink and a stable monohydride step at equilibrium. The hydrogen atoms are gray and the silicon atoms are black.
Table 1Site specific rates of ammonium fluoride etching of Si(111) surfacesa
Because of the anisotropic nature of the ammonium fluoride etching of silicon, it is not possible to produce atomically flat Si(100)–H surfaces by chemical etching. In fact, chemical etching of Si(100) surfaces tends to roughen the surface by exposing Si(111) facets leading ultimately to the formation of porous silicon.16 Under some conditions it is possible to produce relatively flat surfaces with small atomically resolved domains17 but the structure of hydrogenated surfaces produced in this way are not uniform and contain some SiH and SiH3 groups but are predominantly SiH2.18
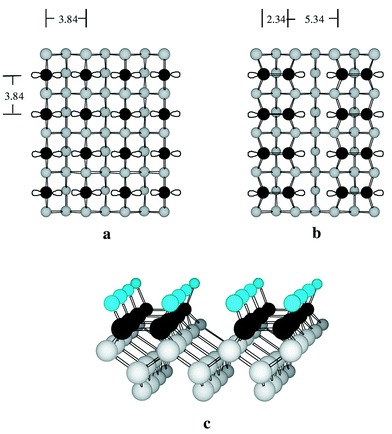
Under UHV conditions it is possible to produce uniform Si(100)–H surfaces. These are produced by first preparing “clean” Si(100) surfaces (i.e. the surface atoms are not chemically terminated but have so-called dangling bonds protruding into space) followed by exposure to hydrogen atoms. Using this approach the surface produced is one which has undergone a reconstruction to form rows of Si–Si dimers (Fig. 4). The reconstruction of the surface to this 2 × 1 structure (which denotes the unit cell dimensions in relation to the unreconstructed surface) is driven by the formation of Si–Si bonds which reduces the number of dangling bonds on the surface atoms from two per silicon to only one. Thus the originally square array of silicon atoms with interatomic separations of 3.84 Å forms an anisotropic surface in which the separation of dimers in the silicon dimer row direction is still 3.84 Å, the separation
of silicon atoms within a dimer is 2.34 Å and the separation between dimers is 7.68 Å from the centres of the bonds or, 5.34 Å between silicon atoms on either edge. The hydrogen terminated surface retains this anisotropic structure which, as will be seen below, has interesting chemical consequences.
Fig. 4 (a) View of the Si(100) unreconstructed surface with two dangling bonds per silicon atom. (b) The 2 × 1 reconstruction of the clean surface with one dangling bond per silicon. (c) View of the hydrogen terminated Si(100) surface. The surface silicon atoms are shown in black and the subsurface atoms in gray for easy viewing.
2.3 Stability of hydrogen terminated surfaces
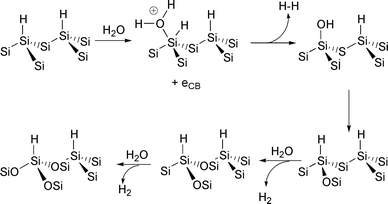
The hydrogen terminated silicon surfaces are actually quite stable and can be handled in air for several minutes before measurable oxidation of the surface occurs. Infrared studies19 have shown that the half-life of the Si–H stretch in air is humidity dependent. The mechanism for the oxidation by water is shown in Fig. 5. In this reaction, water is believed to react with the surface by the donation of an electron to form a pentavalent silicon which subsequently loses dihydrogen to form an Si–OH group. The OH then inserts into an Si–Si back bond to regenerate a surface Si–H bond. The reaction proceeds until all of the back bonds are oxidized. Interestingly, the (SiO)3Si–H species is kinetically stable and is the predominant species on the surface after several hours in humid air. The evidence for this is the observation of the growth of an IR absorption at 2250 cm−1
at the expense of the (Si)3Si–H stretch at 2084 cm−1.19 The implication is that the reaction of water with the (SiO)3SiH species is kinetically limited although a clear explanation of why this should be was not offered.
Fig. 5 Mechanism of oxidation of Si(111)H by water.
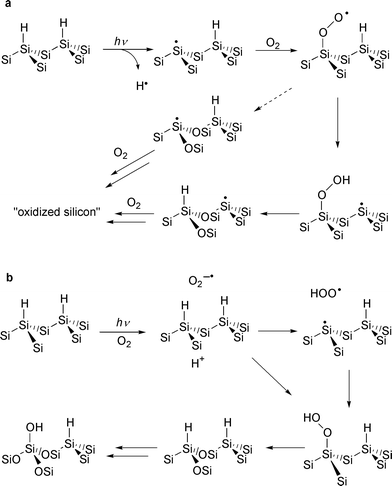
The Si–H surface has been shown to oxidize under photochemical conditions (λ
= 254 nm) in a reaction which presumably involves dioxygen. There are several possible mechanisms for this reaction. Chidsey proposed a mechanism in which irradiation of the surface produces silyl radicals (dangling bonds) on the surface which then react with dioxygen in a surface chain process (Fig. 6a). While this mechanism is plausible, Chatgilialoglu and coworkers 20 found that silylperoxyl radicals undergo a double insertion reaction to insert its oxygens into Si–Si back bonds of silane polymers (Fig. 6a, dotted arrow). One other mechanism should be considered. It is difficult to rule out the formation of superoxide since 254 nm is close to the photoemission threshold of silicon (Fig. 6b). Superoxide was shown by Chidsey to initiate etch pit formation
during the aqueous etching of Si(111).5 The same initiation process in air may lead to surface oxidation. Whatever the mechanism or mechanisms, it is noteworthy that, unlike the oxidation by water, photooxidation leads to a predominantly hydroxylated silicon surface.21
Fig. 6 Two of the possible mechanisms for the photooxidation of Si(111)–H.
3 Reactions of hydrogen terminated silicon
3.1 Formation of Si–C bonds by reaction with alkenes
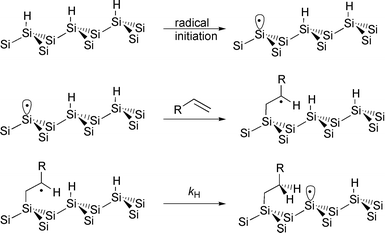
Much of the research in the chemistry of hydrogen terminated silicon surfaces was motivated by the seminal work of Chidsey and his coworkers22,23 who observed the formation of monolayer thick films when Si(111)–H surfaces reacted with alkenes in a formal hydrosilylation reaction in the presence of dibenzoyl peroxide. Based on analogies with radical chain processes of organosilane molecules,24 Chidsey proposed a surface propagated radical chain mechanism (Fig. 7). In this mechanism surface bound silyl radicals (or dangling bonds) are proposed to be formed from the decomposition of radical precursors such as diacyl peroxides or from the irradiation of the surface with UV light.25,26 The silyl radical then reacts with an alkene present either neat or in solution to form a secondary alkyl radical which may then abstract hydrogen from a vicinal SiH
bond. The hydrogen atom transfer reaction provides the saturated organic at the surface and generates a new surface silyl radical which can then continue to propagate the chain. Although not specifically stated, termination of this surface chain process is expected to occur by hydrogen transfer from a molecule in solution or by a radical–radical reaction. There are other constraints on the chain reaction which are discussed below.
Fig. 7 Chisdey mechanism for the radical chain driven reaction of Si(111)–H with alkenes.
Before discussing the mechanism further, it is useful to review briefly some of the physical and chemical properties of these films. One of the challenges in surface chemistry is the analysis of the outcome of a reaction since the number of molecules on the surface tends to be on the order of 10−10 mol cm−2 and products cannot be separated from by-products or contaminants (in contrast to solution chemistry). This makes the interpretation of anomalous behaviour difficult as one can never really rule out the possibility of small amounts of impurities being the dominant influence in a physical measurement. For example, the ability to form highly ordered alkyl films is critically dependent on the purity of the organic molecule forming the monolayer.27
The most common spectroscopic method used to analyse Si–alkyl films is infrared absorption. Normally this is carried out using a geometry referred to as attenuated total internal reflection (ATR) which provides multiple reflection in order to increase the signal-to-noise ratio. Infrared absorption, especially in the alkyl stretch region provides some insights into the extent of molecular ordering of methylene chains.28 The asymmetric methylene stretch, νa, of disordered aliphatic liquids typically are near 2925 cm−1 while for crystalline materials it may be as low as 2915 cm−1. The shift in νa to higher frequency absorption is associated with an increase in the number of gauche defects in the alkyl chains. Highly ordered films tend to adopt an all trans geometry of the methylenes. Chidsey,23 as well as others29–31
find that νa is chain length dependent and varies from values near 2920 cm−1 for a ten carbon chain to values of 2916 cm−1 for a sixteen carbon chain. The implication is that the long alkyl chains form ordered monolayers, at least from the point of view of chain dynamics and conformation. This does not necessarily imply the absence of voids and pinholes in a film.
In the orginal studies of Chidsey and his coworkers the alkenes were used neat. Sieval et al. reported that it is possible to work in solution with dilute alkene but that the choice of solvent has an influence on the molecular ordering of the alkyl monolayers.32 In this study, contact angle measurements (water) were used as an indication of the hydrophobicity of the film. The underlying assumption is that defects in the film due to voids or partial oxidation of the silicon result in a decrease in the contact angle. They found that mesitylene was the only solvent in the study which provided well ordered monolayers and suggested that it is too large to fit in pinholes in the forming film and thus, cannot interfere with the monolayer formation process.
We turn now to the mechanism of the non-catalytic reaction of alkenes with the Si(111)–H surface. Of initial concern was evidence for the formation of an Si–C bond. Typically, it is difficult to remove the last several monolayers of high boiling hydrocarbons from surfaces due to strong physisorption. The most convincing evidence for the formation of an Si–C bond would be the observation of the expected vibrational transition near 780 cm−1. Unfortunately, absorption of bulk silicon below about 1400 cm−1 renders this region in the spectrum inaccessible. In their original work Chidsey and workers23 used somewhat indirect means to imply the formation of a covalent bond. They showed that the surface could withstand exposure to boiling water, boiling chloroform and sonication in dichloromethane. A physisorbed film of hexadecane (for example) would not survive these treatments.
The first direct evidence for the formation of an Si–C bond in a reaction of an alkene with Si(111)–H was reported by Chidsey and coworkers. 26 Using an elegant photoelectron diffraction method they determined the Si–C bond length at this surface to be 1.85 ± 0.05 Å. Sung et al.33 later reported further direct evidence for the Si–C link. Using electron energy loss spectroscopy (EELS) to study the thermal stablility of Si(111)–alkyl and Si(100)–alkyl monolayers, a vibrational transition near 780 cm−1 was clearly observed. The disappearance of this absorption above 615 K and the concomitant reappearance of the Si–H stretch implies that the thermal desorption of the alkyl group is the retro-silylation process resulting in the formation of the alkene and the Si(111)–H surface. It is possible that the cleavage of a single Si–C bond to
form a silicon dangling bond may result in the radical chain “unzipping” of the monolayer from the surface (i.e. the reverse of Fig. 7).
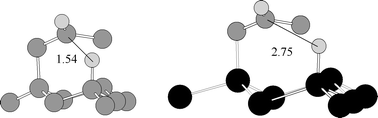
Given the compelling evidence for the formation of the Si–C bond, there still remain some questions regarding the efficacy of the radical mechanism. While the 1,5-hydrogen atom transfer on the surface appears to be well founded in the organic chemistry literature, on closer inspection it is obvious that the increased length of the Si–Si bond compared to a C–C bond imposed greater stereoelectronic constraints on the surface reaction (Fig. 8). Bateman et al. studied the reaction of Si(111)–D with alkenes. The implication of the Chidsey mechanism is that a C–D bond should be uniquely formed at C2 of the original alkene.34 They were unable to detect a C–D stretch and concluded that the radical mechanism as proposed was not correct, at least under the conditions used (initiated by refluxing toluene). A few points should be made about this study. It is possible that the mechanism
of the reaction may depend on the method of initiation. Activated processes such as hydrogen atom donation to an alkyl radical from toluene may occur by a minority pathway when the reactions are initiated at room temperature using photochemical or free radical processes. In refluxing toluene, hydrogen atom transfer from the solvent should be more efficient. Furthermore, there will be a primary deuterium isotope effect on the surface deuterium atom transfer which can slow down the surface process by as much as an order of magnitude. For these reasons, the authors themselves were somewhat equivocal in their conclusions.
Fig. 8 Comparison of a 1,5-hydrogen atom abstraction in a carbon framework compared to a silicon surface. The distances are in ångström. The silicon atoms are black, the carbon atoms are gray and the hydrogen atoms light gray.
Evidence for the surface chain reaction of alkenes with hydrogen terminated silicon surfaces recently was reported by Lopinski et al.35 in an experiment which used the Si(100)–H 2 × 1 surface (Fig. 4) in a UHV environment. Isolated dangling bonds (Fig. 9a) created using an STM tip using a literature method36 were exposed to low vapour pressures (10−8 Torr) of alkenes. After a 3 L exposure to styrene (L = 1 langmuir = 10−6 Torr s defines the number of collisions to form a monolayer if every collision leads to a reaction), some unreacted dangling bonds remained (imaged as ∼1 Å high protrusions). However, at other sites the emergence of distinctly larger protrusions ranging in height from 2 to 3 Å as well as the development of some short lines extending over 2 or 3 surface sites as
seen in Fig. 9b gave an indication of the surface chain process. Upon further exposure to styrene the lines, which tend to appear centered between dimer rows, continued to grow (Fig. 9c). These observations are consistent with the Chidsey radical chain mechanism. The interesting consequence of the anisotropic nature of the Si(100) surface is the preferential growth along one edge of the silicon dimer row. This leads to the growth of self-directed lines and has been proposed to be potentially useful to grow interconnections between molecular scale devices.35 The longest line in Fig. 9d is ∼130 Å, corresponding to 34 adsorption sites. Growth of the lines was observed to stop at pre-existing “missing dimer” or other defects. It is interesting that while there is a clear preference for lines to grow along one edge of a dimer row occasionally double
lines are observed (Fig. 10) suggesting that hydrogen atom abstraction across a dimer row also is feasible. Abstraction across a single dimer has not been observed and is likely to be a prohibitively strained process.
Fig. 9 Sequence of STM images (250 Å
× 140 Å, −2.1 V, 47 pA) of a H-terminated Si(100) surface with a dilute concentration of single dangling bonds upon increasing exposure to (a) 3 L, (b) 28 L, (c) 50 L, (d) 105 L of styrene. The white arrows denote two particular dangling bond sites which lead to the growth of long styrene lines. The missing dimer defect (M) marked in the figure terminates the growth of the line in the top left hand corner of the image (from ref. 35).
Fig. 10 (a) An STM image (350 Å2, −3.3 V, 10 pA) of an Si(100)–H surface with a dilute concentration of single Si dangling bonds after exposure to 200 L of styrene. Examples of single (S) and double lines (D) are indicated. (b) A molecularly resolved STM image (90 Å2, −2.2 V, 470 pA) of a single styrene line. The features in the line are spaced by ∼3.8 Å. The inset depicts a possible conformation of a 5 molecule chain of adsorbed styrene molecules, each of which is covalently bound to a single Si dimer (from ref. 35).
The observation of line growth on the Si(100)–H 2 × 1 surface while not direct evidence, is certainly strong indirect evidence of the efficacy of the Chidsey mechanism on Si(111). Even though the Si–H bonds form an angle of about 110° with respect to the surface, the relative positioning of adjacent Si–H bonds (3.84 Å) is the same. Unlike the Si(100)–H 2 × 1 surface, Si(111)–H is a three-fold symmetric surface so the radical chain process will not propagate along a single direction but instead should propagate as a random walk on the surface. Using Monte Carlo simulation methods it was predicted that there should be on average about 80 steps in the radical chain process.37 The hypothesis was recently tested using an STM approach similar to that used for the reaction on the Si(100)–H surface.38 In this case, the Si(111)–H surface was prepared by chemical etching
and introduced into a UHV chamber. Dangling bonds were generated using the STM tip and the surfaces were exposed to styrene. The resulting STM images (Fig. 11) were consistent with the prediction made by Chidsey. Each original dangling bond in Fig. 11a reacted with styrene to form an island of modified silicon with an average diameter of about 2 nm (Fig. 11b). The growth of these islands was self-limited and continued exposure to styrene did not lead to observable changes. The average chain length of 25 which can be deduced from the images, is about a factor of three smaller than that predicted by the simulation. While the simulation assumed all sites were equally reactive, it would have been reasonable to predict much longer chains since steric effects might inhibit reactions of surface sites adjacent to other reacted sites. In fact, the opposite seems to be true suggesting that attractive interactions
tend to pull the chain reaction back on itself.
Fig. 11 Occupied state STM images (215 Å
× 130 Å, −2.1 V, 44 pA) of an H-terminated Si(111) surface with isolated dangling bonds created by desorption activated with the STM tip. (a) before dosing with styrene and, (b) after exposure to 12 L of styrene. The black dots in (b) mark the positions of the initial dangling bonds, showing that these sites serve to nucleate the growth of styrene islands (from ref. 38).
It should be made clear that the demonstration of the chain reaction under UHV conditions is not in itself proof that the alkene reactions proceed by a radical chain mechanism. While it has generally been assumed, it is not proven that the thermal, photochemical and radical initiated reactions have a common intermediate. Clearly this is an area which requires more careful examination using both UHV approaches as well as the more established methods used by free radical chemists such as free radical clocks and other mechanistic probes.39
One interesting observation in the UHV study on the Si(100) surface35 was that isolated reactions occurred but lines did not form on exposure to more than 200 L of propylene. It is interesting to consider these reactions from the point of view of known free radical kinetics and thermodynamics. Some relevant rate constants for the reactions of the (TMS)3Si˙ radical are shown in Table 2. The most simple explanation for the inability of propylene to react is that only a small fraction of collisions lead to the formation of the carbon centered radical intermediate. From the rate constants in Table 2, the barrier for the addition of (TMS)3Si˙ to propylene and styrene can be estimated to be about 4 and 6 kcal mol−1, respectively (the rate constant for reaction of a silyl radical with an n-alkene is about 100 times slower than the corresponding
reaction with styrene). The reaction of propylene with a dangling bond is estimated to be exothermic by about 9 kcal mol−1 while the hydrogen atom abstraction by the alkyl radical is exothermic by an additional 16 kcal mol−1.35 For styrene the addition and hydrogen abstraction steps are exothermic by 18 and 3 kcal mol−1, respectively. Thus, for propylene the rate constant for desorption of the intermediate radical will be 102 s−1
≤
kd
≤ 107 s−1 compared to 10−3 s−1
≤
kd
≤ 1 s−1 for the styrene reaction (the upper limits assume no barrier for the addition reaction). The observation of a line 34 steps long after only ca. 100 L exposure implies that the sticking coefficient is high. The sticking coefficient, which is a measure of the number of
collisions per site that lead to a reaction is likely to be overestimated by this simple method because there will be a reaction radius around each dangling bond from which physisorbed molecules can diffuse to the reaction site before desorption. This radius depends on the relative barriers for reaction compared to desorption of the physisorbed species. The high sticking coefficient implies a low barrier to addition of the alkene to the dangling bond so the upper limit for reactivity is more reasonable. Thus, it is only required that the rate of the hydrogen atom transfer is comparable to the rate of desorption of the intermediate radical, kH > 1 s−1. If the relative rates of hydrogen atom transfer on the surface and in solution are similar then a lower limit of kH > 104 s−1 can be set for the reaction of propylene.
Table 2Rate constants for reactions of the tris(trimethylsilyl)silyl radical at 25 °Ca
Reaction
Rate constant/M−1 s−1
aFrom ref. 24.bEstimated assuming half of the change in the driving force appears at the transition state.
One additional complication arises when this reasoning is extended to the solution reaction. The pseudo first order polymerization rate constant, kP, of the surface radical with neat styrene in solution should be 9 M × 50 M−1 s−1
≈ 400 s−1. Since polymer is not detected during the surface process (even when the reactions are carried out in neat stryrene)37 this puts a further constraint on the lower limit of kH which must be much greater than the rate of polymerization; kH > 105 s−1 for styrene. This value cannot be directly compared to the rate constants in Table 2 since those values are for bimolecular processes. The equivalent rate constant in a unimolecular process should be 3–4 orders of magnitude greater (assuming the entropy changes are about the same). This
lower limit on the surface reaction raises an interesting conundrum since it suggests the barriers for hydrogen atom transfer in the styrene and propylene reactions are about the same as those in the corresponding bimolecular processes in which the transition states are essentially unstrained. This seems unlikely in view of the large separation between the radical center and the Si–H bond (Fig. 8). This leaves the interesting prospect that not even relative rate constants from solution reactions of radicals can be applied to surface reactions or that there are competing mechanisms in the solution reaction of styrene with the Si(111)–H surface. In either case, this is fertile ground for the physical organic chemist.
While the mechanism of formation of these alkyl monolayers is important, the structure of the film is equally important if any of the proposed applications are to be realized. Since the distance between silicon atoms on the Si(111) surface is 3.84 Å and the diameter of an alkyl chain is about 4.2 Å it is clear that complete 1 × 1 coverage of the silicon surface is not possible.23 The thickness of the film which can measured by XPS,40 ellipsometry,23 or X-ray reflectivity23,31 provides a measure of the average tilt angle of the axis of the hydrocarbon chain with respect to the surface. These various measurements provide a tilt angle of about 30 ± 5° from the surface normal for the product from reaction of hexadecene which is consistent with a surface coverage of about 50 percent. Recently, Sieval et al.41,42
developed molecular modeling methods which allow some structural parameters to be estimated. The calculations were able to reproduce experimentally determined tilt angles and confirmed that from an energetic viewpoint the optimal coverage of hexadecyl groups on the Si(111) surface was 50 percent. One difficulty in comparing computational results with experiment is the method used to define the starting surface. Sieval et al. used high symmetry monolayers for their calculations. In reality, it is unlikely that such high symmetry can be achieved. If the reaction proceeds by a radical mechanism then small domains of alkyl islands are expected. On the other hand, if the reactions occur randomly on the surface then at best only small ordered domains are expected. Nevertheless, the computational methodology developed by Sieval et al. is important as it demonstrates that edge effects in finite surface calculations can be overcome using periodic boundary conditions. The calculations
do provide valuable insights and can be applied, in principle, to any arbitrary starting structure.
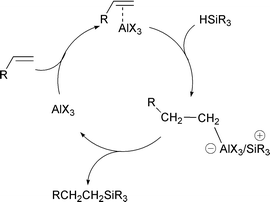
At least one clearly non-radical route to the formation of Si–C bonds with alkenes has been reported.29 Hydrosilylation of alkenes by Lewis acids or transition metals is well known in the molecular organosilane literature. Buriak and coworkers43,44 showed that the surface of hydrogen terminated porous silicon could effectively be hydrosilylated in a room temperature reaction using EtAlCl2 as the catalyst. The mechanism of this reaction is shown in Fig. 12. Although it is possible to catalyse the reaction of an alkene on the Si(111)–H surface,29 the requirement to bring the catalyst in close proximity to the surface raises concerns about steric effects. These Si(111)–R surfaces were shown to have only about 80 percent of the maximum coverage achieved using the photochemical approach.
Fig. 12 Mechanism of the Lewis acid catalysed reaction of alkenes with the Si(111)–H surface.
3.2 Formation of Si–C bonds by other reactions
3.2.1 Grignard and alkyllithium reactions. The analogy drawn between the radical reactions of organosilanes and those of the Si(111) surface led to the exploration of a number of reactions to establish the scope of the surface reactions. Bansal et al.45 showed that it was possible to chlorinate the Si(111)–H surface in a benzene solution of PCl5 and then to react that surface with an organic Grignard reagent (reactions (3) and (4)). Since then a number of halogenation methods have been developed. Chidsey and coworkers25 showed that a light stimulated reaction of molecular chlorine or bromine led to the formation of the corresponding halogenated surface by a classic radical process in just a few seconds (i.e. chlorine atom abstraction of a hydrogen atom followed by chlorine atom transfer to the surface from molecular chlorine). He et
al.46 later showed that a variety of halogenating reagents (N-bromosuccinimide, bromotrichloromethane, carbon tetrachloride) could be used to provide Si(111)–halogen surfaces. These halogenated surfaces all react with carbanion equivalents in formal substitution reactions.
(3)
Si(111)Cl + RMgBr → Si(111)R + ClMgBr
(4)
In their study of the reactions of Grignard reagents with halogenated surfaces, Boukherroub et al. were surprised to find that similar monolayer films were formed by the direct reaction of a Grignard reagent with the Si(111)–H surface.29 The reaction was particularly surprising since there was not a precedent in the molecular organosilane literature. Monolayers formed in this direct reaction have similar spectroscopic,29 chemical29 and electrochemical properties47,48 to those formed in the two step process.49,50 A mechanism for the formation of an Si–C bond under these conditions was not suggested. However, at the end of this review we will take a closer look at the molecular analogy and offer some speculation about reactions which appear anomalous.
3.2.2 Electrochemical formation of monolayers. Allongue et al. reported the reaction of aryl radicals, formed by the electroreduction of aryl diazonium salts, to form a covalently modified highly ordered pyrolytic graphite surface.51 The key step in this reaction was the reduction of an arenediazonium salt to form an aryl radical which then directly bonded to the graphite surface. This idea was extended to reactions with the Si(111)–H surface (reactions (5)–(7)).52,53 One of the advantages in the use of arenediazoniums is the compatibility with a number of substituents including Br, NO2, CN, NH2, COOH and alkyl providing excellent synthetic flexibility. In addition, these reactions produce highly ordered domains and provided the first STM view of the predicted 2 × 1 organic structure on the Si(111) surface
(Fig. 13).53
ArN2+
+ e−
→ Ar˙
+ N2
(5)
Ar˙
+ Si(111)H → ArH + Si(111)˙
(6)
Ar˙
+ Si(111)˙
→ Si(111)–Ar
(7)
Fig. 13 Atomically resolved STM image (5 × 5 nm2) of (a) the 1 × 1 Si(111)–H surface and (b) the Si(111)–C6H4Br surface formed by the electrochemical reduction of 4-bromobenzenediazonium tetrafluoroborate showing the 2 × 1 structure of the organic film (images courtesy of P. Allongue, ref. 53). The unit cells are shown in each image.
Oxidation processes also can lead to the formation of radicals near the Si(111)–H surface. Recently Chazalviel and coworkers54 used the electrochemical oxidation of methyl magnesium iodide to form a fully methylated Si(111) surface. Methyl is the only alkyl group which should be able to form a relatively stress free 1 × 1 layer on this surface. The work was stimulated by their previous studies of electrochemical modification of porous silicon with Grignard reagents.55 While the mechanism of the electrochemical process is a subject of speculation, the most simple mechanism is one in which CH3˙ and/or I˙ are formed by the electrochemical degradation of the Grignard reagent.
3.3 Formation of Si–O bonds
Cleland et al.56 reported that alcohols react with Si(111)–H surfaces to form directly Si(111)–OR modified surfaces. These direct reactions are synthetically flexible and allow a wide range of surface function to be incorporated into the monolayer films. Of particular interest was the reaction of ferrocenylmethanol which provided an electroactive monolayer on the silicon surface (reactions (8) and (9)). Electrochemical characterization of these monolayer films56,57 allowed the surface coverage to be estimated to be 1 × 10−10 mol cm−2 which represents about 10 percent coverage on a per silicon basis.
Si(111)–H + FcCH2OH → Si(111)–OCH2Fc
(8)
Si(111)–OCH2Fc ⇌ Si(111)–OCH2Fc+˙
+ e−
(9)
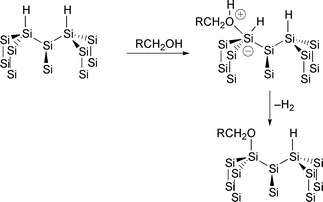
The mechanism of formation and properties of the Si(111)–OR surfaces formed by the reaction of alcohols and aldehydes with the Si(111)–H surface has been studied in more detail by Boukherroub et al. using FTIR, XPS and atomic force microscopy.58 The mechanism proposed for the reaction of alcohols (Fig. 14) is similar to the oxidation of silicon by water.59 The reaction proceeds by the attack of the alcohol on a terrace site followed by loss of dihydrogen from the pentacoordinate silicon. Unlike water, alcohols do not etch the silicon surface. Hines and coworkers showed that even small amounts of propan-2-ol inhibit step-flow etching; the rate of fluoride etching of an Si(111)–OCHMe2 kink site is 5 orders of magnitude slower than reaction with the hydroxylated site.60 Boukherroub et al. also showed that surfaces containing
significant etch pits result if traces of water are not removed from the alcohol reactions (Fig. 15a). One simple approach is to add a chemical reagent such as trimethylsilyl chloride which reacts with traces of water (Fig. 15b).
Fig. 14 Mechanism of reaction of alcohols with Si(111)–H surfaces.
Fig. 15 Contact AFM images from ref. 58 (3 × 3 μm2) of Si(111)–OC10H21 formed by the thermal reaction of (a) neat decanol and (b) decanol containing 5 percent v/v chlorotrimethylsilane. In the absence of water there are few etch pits on the surface.
Effenberger et al.30 reported the formation of Si(111)–OR monolayers from the photochemical reaction of aldehydes. In this study, it was shown that the photochemical process could be used to chemically pattern the surface on the micron scale by exposing a thin film of the organic molecule on a freshly etched shard of Si(111)–H to UV light through a mask. The irradiated regions reacted with the aldehyde. Effenberger et al. showed that there was a wavelength dependence on the efficiency of the reaction with a maximum near 380 nm. This is in contrast to the report by Cicero and Chidsey37 who found that the reaction efficiency increased monotonically with decreasing wavelength up to 254 nm. The reasons for this discrepancy are not clear.
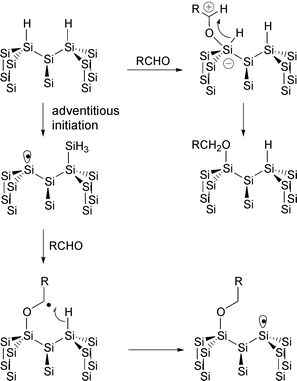
Boukerroub et al. found that like alkenes, there is a thermal reaction of aldehydes with the Si(111)–H surface.58 However, these reactions occur at much lower temperatures than the corresponding alkenes. Two possibly competing mechanisms were proposed (Fig. 16). The first involves attack of the carbonyl group on the silicon surface followed by a formal 1,2-hydride shift. The second is similar to the radical chain reaction of alkenes (see above). In practice, it is difficult to distinguish these mechanisms.
Fig. 16 Mechanism of reaction of aldehydes with the Si(111)–H surface.
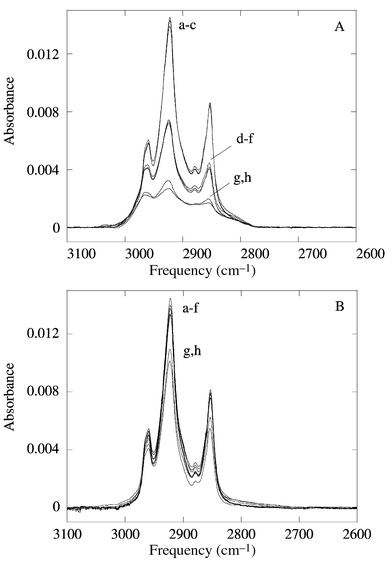
Unlike in the case of alkenes, there is no direct evidence for the formation of the Si–O bond. The high chemical stability as measured by FTIR has been used as indirect evidence for a covalently modified surface. One interesting difference between the aldehyde modified surfaces and the alcohol modified surfaces is seen in their respective chemical stability. While the alcohol modified surface is stable under standard degreasing conditions such as rinsing with trichloroethane, boiling in chloroform or sonicating in dichloromethane, it was found to be unstable in boiling water, a process which is known to etch silicon at defects in the monolayer (Fig. 17A). The aldehyde modified surfaces are stable under these conditions and only are removed by exposure to HF (Fig. 17B). A similar lack of stability was found for surfaces formed by the reaction of alcohols with Si(111)–Cl which should form a similar Si(111)–OR
surface.61
Fig. 17 Infrared spectra of the C–H stretch region of the alcohol (A) and aldehyde (B) modified surfaces. The surfaces were subjected to the following treatments: (a) rinsed with trichloroethane, (b) sonicated in dichloromethane for 5 min, (c) immersed in boiling chloroform for 1 h, (d) immersed in boiling water for 1 h, (e) immersed in 1.2 M HCl at 25 °C for 1 h, (f) immersed in water at 25 °C for 16 h, (g) immersed in 2 percent HF for 2 min, (h) immersed in 2 percent HF for 10 min (from ref. 58).
4 Some comments on molecular analogies
Much of the work on the modification of hydrogen terminated silicon surfaces has been motivated by molecular analogies. Indeed, the original proposal of a surface radical chain reaction and various nucleophilic substitution reactions of silyl halides have close analogies to molecular organosilane reactions. However, there are a number of reactions for which no molecular counterpart exists. In particular, the direct thermal reactions of Grignard reagents and alcohols with model systems such as (TMS)3SiH do not lead to substitution of the Si–H bond but lead to the cleavage of Si–Si bonds. The thermal silylation of aldehydes has been reported but, in general, much higher temperatures or catalysts are required.62 So what is different about these surfaces compared to the simple molecular model compounds? There is a tendency to focus on the local bonding in these systems. The Si–H bond in (TMS)3Si–H and Si(111)–H
seem similar and it is deceptively simple to focus on that aspect which is so well understood by physical organic and synthetic chemists. However, there is one key difference between the molecule and the material. Silicon has a band gap of about 1.1 eV while the HOMO–LUMO gap in the corresponding molecules are 8–11 eV. This has consequences for the reactions of nucleophilic and electrophilic species where frontier orbital interactions determine the reactivity and selectivity of the reaction.
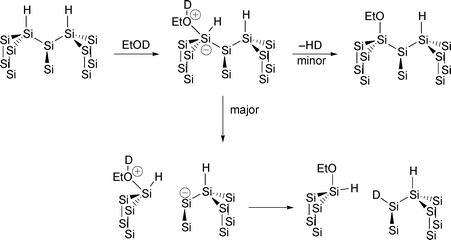
The comparison of the reactions of Si(111)–H and porous silicon serves to illustrate this point. As seen above, alcohols react with the Si(111)–H surface in a mechanism which is believed to be initiated by the interaction of a lone pair of electrons on oxygen with the states in the conduction band of silicon (electron injection). This is followed by the loss of molecular hydrogen (Fig. 14). Whether the pentavalent silicon is an intermediate or transition state is not of importance for this discussion. In the end, the result is the formation of an Si–OR bond on the intact single crystal surface. In contrast, Kim and Laibinis63 showed that the reaction of ethanol with porous silicon results in the cleavage of Si–Si back bonds (Fig. 18). This was demonstrated using EtOD and showing the appearance of new Si–D bonds concomitant with the formation of Si–OEt bonds
using FTIR.
Fig. 18 Mechanism of reaction of ethanol (OD) with porous silicon.
It is interesting to speculate about the differences between these reactions. While the structure of the bulk material is the same for both substrates, the nanoscale structure of porous silicon increases the band gap as a result of quantum confinement.16 It is likely that this is the factor which determines the course of the chemical reaction. Wide band gap silicon structures such as those in porous silicon and organosilane molecules inhibit electron injection or nucleophilic charge donation allowing other processes such as bond cleavage to dominate.
A similar mechanism involving the cleavage of Si–Si bonds was proposed for the reaction of Grignard reagents with porous silicon.64 However, the oxidation potentials of primary carbanions (ca.
−1.5 V vs. SCE or about 3.2 eV)65 are low enough that direct electron injection into the conduction band of crystalline silicon (4 eV) is feasible. Thus, it is likely that a key step in the reaction of Grignard reagents with Si(111)–H is the formation of alkyl radicals at the interface. The formation of Si–C bonds may be by radical–radical coupling or by reaction of a carbanion equivalent with a silicon dangling bond in a reaction related to the SRN1 process (reactions (10)–(13)).66 While his mechanism should be considered to be speculative at this time, it is analogous to the electrochemical
reactions studied by Chazalviel and coworkers.54 One difficulty with the reactions as shown is the requirement that charge should accumulate in the material (i.e. two electrons injected per Si–C bond formed); a problem not encountered with the electrochemical process.
RMgBr → R˙
+ MgBr+
+ eCB
(10)
R˙
+ Si(111)H → RH + Si(111)˙
(11)
R˙
+ Si(111)˙
→ Si(111)R
(12)
RMgBr + Si(111)˙
→ Si(111)R + MgBr+
+ eCB
(13)
More work is necessary to fully understand the mechanism of the Grignard reaction. However, reactivity trends of Si(111)–H and (TMS)3Si–H or porous silicon suggest that frontier orbital interactions which require electron donation into the LUMO or conduction band of the silicon species are important in the reactions of nucleophilic species.
5 Functionalization of monolayers
In most of the examples above, the reactions lead to organic monolayer films that are terminated with a methyl group. While this has been useful for mechanistic studies as well as for passivation and chemical stabilization, the low reactivity of the terminal methyl group makes further manipulation of the surface physical or chemical properties difficult. The incorporation of biocomponents such as DNA or proteins onto these surfaces for the construction of integrated biosensors and microarrays will require the distance (at least average distance) between reactive centers to be carefully controlled in order to reduce steric interference while at the same time optimizing the sensitivity of a device.67
The possibility of creating a functionalized organic film on a single crystal surface has been demonstrated23,44,52,53,68,69 but only in a few cases have the sequential reactions of the organic film been carried out.31,70–72 The ability to incorporate more complex organic or bioorganic structures at the interface means new strategies should allow the chemical functionality of the surface to be tailored to meet predefined physical or chemical requirements.
Chidsey and coworkers addressed this problem using a two step process in which the surface is first chlorosulfonated by a photoinitiated free radical reaction followed by sulfonamide formation by reaction of the sulfonyl chloride with an amine (reactions (14) and (15))72 or by reaction with photogenerated singlet carbenes.71 Using this approach it was possible to functionalize the surface with a diverse range of amines including those containing DNA and dendrimers. In both of these reactions, the extent of chemical reaction was difficult to control as it depends on a number of factors including the concentration of reagents and the intensity and spectrum of the light source.
Sieval et al. carried out more traditional solid phase synthetic procedures from modified surfaces.31 Thermal reaction of alkenes terminated with ester groups reacted with the Si(100)–H to form an ester terminated surface which could be hydrolyzed to form a carboxylic acid modified surface or reduced with LiAlH4 to form an alcohol modified surface (reactions (16) and (17)). The carboxylic acid surface could be re-esterified by refluxing the surface in an acidified solution of a different alcohol.
(16)
(17)
While this demonstrated the possibility of carrying out sequential chemical transformations on the surface, a poor signal to noise ratio in the carbonyl region made it difficult to assess the overall chemical yields and, perhaps more importantly, the reaction conditions were too harsh to be compatible with most biological materials. Boukherroub and Wayner further elaborated this approach to the functionalization of the covalently bonded alkyl monolayers by showing that reactions commonly used in solid phase synthesis could be adapted to the silicon surface chemistry.70 The photochemical reaction of ethyl undecylenate with Si(111)–H provided a surface whose chemical properties were manipulated using standard chemical and solid phase chemical procedures (reactions (18)–(21)). The surface density of the reactive esters could be controlled simply by dilution of the ester with an n-alkene.
(18)
(19)
(20)
(21)
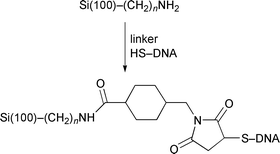
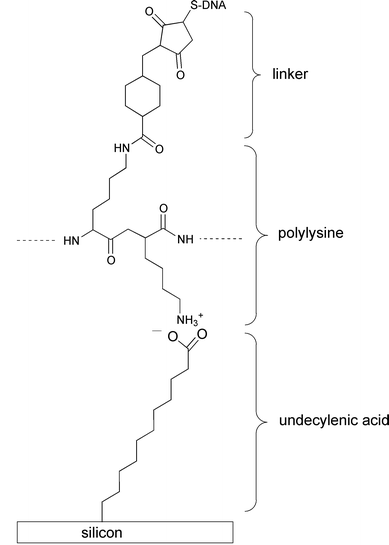
Strother and coworkers73,74 modified silicon surfaces with DNA using two different chemical approaches. In one approach they hydrolyzed a monolayer of Si(111)–(CH2)10C(O)OCH2CF3 (prepared from the photoinitiated reaction of the corresponding alkene with Si(111)–H) to the undecylenate ion. This served as a substrate for the attachment of DNA by means of an electrostatically adsorbed layer of polylysine and the attachment of thiol-modified DNA using a heterobifunctional cross-linker (Fig. 19).73 The second method used a similar linking method but an amino terminated alkyl chain was formed on the silicon surface by the reaction of t-Boc-protected 10-aminodec-1-ene followed by hydrolysis (reactions (22) and (23)).74 The density of DNA binding sites on the
surface could be controlled using the same method reported by Boukherroub and Wayner.70
(22)
(23)
Fig. 19 Attachment of DNA to modified Si(111) surfaces (ref. 74).
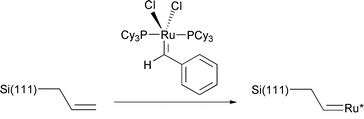
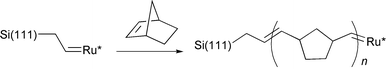
All of the surface chemistry described to this point results in the formation of monolayer films on the silicon surface, typically <2 nm in thickness. Besides being chemically robust, these monolayers provide excellent electronic passivation for the silicon surfaces which may have electrical defect densities of less than one per 50000 surface atoms (seemingly as good as the defect density found in the best semiconductor devices).75 Lewis and coworkers recently described a method using ring opening metathesis polymerization which allows a polymer film of uniform thickness to be grown from an Si(111) surface modified by the reaction of Si(111)–Cl with allyl magnesium bromide.76 Activation with a ruthenium complex allows films as thick as 5 μm to be grown (reactions (24) and (25)). These polymer films may find applications in the fabrication of hybrid organic-silicon molecular
or optical devices.
(24)
(25)
6 Concluding remarks
This review has summarized work carried out in the area of organic modification of hydrogen terminated silicon surfaces. While the tools and instrumentation used in the study of surface reactions may not be familiar to the physical organic chemist, many of the reactions and concepts are the same. Much of the work so far has focused on exploring the scope of the chemistry and on understanding the structure and quality of the organic-silicon interfaces. There has been less of a focus on delineating the mechanisms of formation of Si–alkyl monolayers. The use of techniques such as STM and AFM provide opportunities for physical organic chemists, together with surface scientists, to study the mechanisms and rates of surface processes.
We have alluded within the review to what we believe to be some of the key challenges that require a contribution from physical organic chemists. One of these is to establish kinetics and thermodynamics of the reaction of alkenes with isolated dangling bonds. This includes barriers to addition of alkenes as well as the time scale for the hydrogen atom transfer step in the surface chain reaction. One deceptively simple approach is to use a clock reaction such as the hex-5-en-1-yl rearrangement. However, there are some interesting potential problems when taken to the surface. First and foremost is the analysis of the outcome. Establishing product ratios either by spectroscopy or even STM is not straightforward. In addition, there are as yet untested suppositions that the cyclization is not inhibited on the surface and that the cyclized species will abstract a hydrogen atom from the surface. One key difference between the solution reaction and the surface reaction is that the surface processes
are constrained by the crystallographic periodicity of the surface; a factor which must be considered both in the interpretation of an experimental result and in the design of efficient surface processes.
Another important consideration is the influence of the small band gap of silicon compared to silane molecules on the mechanism of the surface reactions. We have speculated that nucleophilic processes are promoted by donation into the conduction band by analogy with the reaction hydrogen terminated silicon surfaces with water. The use of mechanistic probes should allow the efficacy of some key reactions and intermediates to be tested.
The emergence of free radical chemistry as a useful tool in synthetic chemistry was preceded by many years of fundamental mechanistic and kinetic studies. Similarly, if the potential of hybrid devices based on the convergence of semiconductor and organic materials is to be achieved, it must be underpinned by a detailed understanding of the mechanism and kinetics of the surface reactions.
7 References
Issued as NRCC publication Number 48916.
H. N. Waltenburg and J. T. J. Yates, Chem. Rev., 1995, 95, 1589 CrossRefCAS.
R. A. Wolkow, Ann. Rev. Phys. Chem., 1999, 50, 413 Search PubMed , and references cited therein.
G. S. Higashi, Y. J. Chabal, G. W. Trucks and K. Raghavachari, Appl. Phys. Lett., 1990, 12, 656 CrossRefCAS.
C. P. Wade and C. E. D. Chidsey, Appl. Phys. Lett., 1998, 71, 1679 CrossRefCAS.
P. Allongue, C. Henry de Villeneuve, S. Morin, R. Boukherroub and D. D. M. Wayner, Electrochim. Acta, 2000, 45, 4591 CrossRefCAS.
P. Jakob and Y. J. Chabal, J. Chem. Phys., 1991, 95, 2897 CrossRefCAS.
H. E. Hessel, A. Feltz, M. Reiter, U. Memmert and R. J. Behm, Chem. Phys. Lett., 1991, 186, 275 CrossRefCAS.
P. Allongue and J. Kasparian, Microsc. Microanal. Microstruct., 1994, 5, 257 Search PubMed.
P. Allongue, V. Kieling and H. Gerischer, Electrochim. Acta, 1995, 40, 1353 CrossRefCAS.
Y.-C. Huang, J. Flidr, T. A. Newton and M. A. Hines, Phys. Rev. Lett., 1998, 80, 4462 CrossRefCAS.
J. Flidr, Y.-C. Huang and M. A. Hines, J. Chem. Phys., 1999, 111, 6970 CrossRefCAS.
Y. C. Huang, J. Flidr, T. A. Newton and M. A. Hines, J. Chem. Phys., 1998, 109, 5025 CrossRefCAS.
J. Flidr, Y. C. Huang, T. A. Newton and M. A. Hines, Chem. Phys. Lett., 1999, 302, 85 CrossRefCAS.
H. Luo and C. E. D. Chidsey, Appl. Phys. Lett., 1998, 72, 477 CrossRefCAS.
A. G. Cullis, L. T. Canham and P. D. J. Calcott, J. Appl. Phys., 1997, 82, 909 CrossRefCAS.
K. Arima, K. Endo, T. Kataoka, Y. Oshikane, H. Inoue and Y. Mori, Appl. Phys. Lett., 2000, 76, 463 CrossRefCAS.
Y. J. Chabal, G. S. Higashi, K. Raghavachari and V. A. Burrows, J. Vac. Sci. Technol., A., 1989, 7, 2104 CrossRefCAS.
T. Miura, M. Niwano, D. Shoji and N. Miyamoto, J. Appl. Phys., 1996, 79, 4373 CrossRefCAS.
C. Chatgilialoglu, A. Guerrini, M. Lucarini, G. F. Pedulli, P. Corrozza, G. Da Roit, V. Borzatta and V. Licchini, Organometallics, 1998, 17, 2169 CrossRefCAS.
J. C. T. Wojtyk, R. Boukherroub and D. D. M. Wayner, J. Am. Chem. Soc., 2001, 123, 1535 CrossRefCAS.
M. R. Linford and C. E. D. Chidsey, J. Am. Chem. Soc., 1993, 115, 12631 CrossRefCAS.
M. R. Linford, P. Fenter, P. M. Eisenberger and C. E. D. Chidsey, J. Am. Chem. Soc., 1995, 117, 3145 CrossRefCAS.
C. Chatgilialoglu, Acc. Chem. Res., 1992, 25, 188 CrossRefCAS , and references cited therein.
J. Terry, R. Mo, C. Wigren, R. Cao, G. Mount, P. Pianetta, M. R. Linford and C. E. D. Chidsey, Nucl. Instrum. Methods Phys. Res., Sect. B., 1997, 133, 94 Search PubMed.
J. Terry, M. R. Linford, C. Wirgen, R. Cao, P. Pianetta and C. E. D. Chidsey, Appl. Phys. Lett., 1997, 71, 1056 CrossRefCAS.
A. Badia, R. B. Lennox and L. Reven, Acc. Chem. Res., 2000, 33, 475 CrossRefCAS.
A. Ulman, An Introduction to Ultrathin Organic Films, Academic Press, San Diego, 1991 Search PubMed.
R. Boukherroub, F. Bensebaa, S. Morin and D. D. M. Wayner, Langmuir, 1999, 15, 3831 CrossRefCAS.
F. Effenberger, G. Gotz, B. Bidlingmaier and M. Wezstein, Angew. Chem., Int. Ed., 1998, 37, 2462 CrossRefCAS.
A. B. Sieval, A. L. Demirel, J. W. M. Nissink, M. R. Linford, J. H. v. d. Maas, W. H. d. Jeu, H. Zuilhof and E. J. R. Sudholter, Langmuir, 1998, 14, 1759 CrossRefCAS.
A. B. Sieval, V. Vleeming, H. Zuilhof and E. J. R. Sudholdter, Langmuir, 1999, 15, 8288 CrossRefCAS.
M. M. Sung, G. J. Kluth, O. K. Yauw and R. Maboudian, Langmuir, 1997, 13, 6164 CrossRefCAS.
J. E. Bateman, R. D. Eagling, B. R. Horrocks and A. Houlton, J. Phys. Chem. B., 2000, 104, 5557 CrossRefCAS.
G. P. Lopinski, D. D. M. Wayner and R. A. Wolkow, Nature, 2000, 406, 48 CrossRefCAS.
P. Avouris, R. E. Walkup, A. R. Rossi, H. C. Akpati, P. Norlander, T. C. Shen, G. C. Abeln and J. W. Lyding, Surf. Sci., 1996, 363, 368 CrossRefCAS.
R. L. Cicero and C. E. D. Chidsey, Langmuir, 2000, 16, 5688 CrossRefCAS.
R. L. Cicero, C. E. D. Chidsey, G. P. Lopinksi, D. D. M. Wayner and R. A. Wolkow, Langmuir, 2001 , in the press.
M. Newcomb, Tetrahedron, 1993, 49, 1151 CrossRefCAS.
J. He, Z. Lu, S. A. Mitchell and D. D. M. Wayner, J. Am. Chem. Soc., 1998, 120, 2660 CrossRefCAS.
A. B. Sieval, B. van den Hout, H. Zuilhof and E. J. R. Sudholdter, Langmuir, 2001, 17, 2172 CrossRefCAS.
A. B. Sieval, B. van den Hout, H. Zuilhof and E. J. R. Sudholdter, Langmuir, 2000, 16, 2987 CrossRefCAS.
M. P. Stewart and J. M. Buriak, Adv. Mater., 2000, 12, 859 CrossRefCAS.
J. M. Buriak and M. J. Allen, J.
Am. Chem. Soc., 1998, 120, 1339 CrossRefCAS.
A. Bansal, X. Li, I. Lauerman and N. S. Lewis, J. Am. Chem. Soc., 1996, 118, 7225 CrossRefCAS.
J. He, S. N. Patitsas, K. F. Preston, R. A. Wolkow and D. D. M. Wayner, Chem. Phys. Lett., 1998, 286, 508 CrossRefCAS.
H.-Z. Yu, R. Boukherroub, S. Morin and D. D. M. Wayner, Electrochem. Commun., 2000, 2, 562 CrossRefCAS.
H.-Z. Yu, S. Morin, D. D. M. Wayner, P. Allongue and C. Henry de Villeneuve, J. Phys. Chem. B., 2000, 104, 11157 CrossRefCAS.
A. Bansal and N.
S. Lewis, J. Phys. Chem. B., 1998, 102, 1067 CrossRefCAS.
A. Bansal and N. S. Lewis, J. Phys. Chem. B., 1998, 102, 4058 CrossRefCAS.
P. Allongue, M. Delamar, B. Desbat, O. Fagebaume, R. Hitmi, J. Pinson and J.-M. Saveant, J. Am. Chem. Soc., 1997, 119, 201 CrossRefCAS.
C. Henry de Villeneuve, J. Pinson, M. C. Bernard and P. Allongue, J. Phys. Chem. B., 1997, 101, 2145.
P. Allongue, C. Henry de Villeneuve, J. Pinson, F. Ozanam, J. N. Chazalviel and X. Wallart, Electrochim. Acta, 1998, 43, 2791 CrossRefCAS.
A. Fidelis, F. Ozanam and J.-N. Chazalviel, Surf. Sci., 2000, 444, L7 CrossRefCAS.
T. Dubois, F. Ozanam and J.-N. Chazalviel, Electrochem. Soc. Proc., 1997, 97, 296 Search PubMed.
G. Cleland, B. R. Horrocks and A. Houlton, J. Chem. Soc. Faraday Trans., 1995, 91, 4001 RSC.
R. D. Eagling, J. E. Bateman, N. J. Goodwin, W. Henderson, B. R. Horrocks and A. Houlton, J. Chem. Soc., Dalton Trans., 1998, 1273 RSC.
R. Boukherroub, S. Morin, P. Sharpe and D. D. M. Wayner, Langmuir, 2001 , in the press.
P. Allongue, V. Costa-Kieling and H. Gerischer, J. Electrochem. Soc., 1993, 140, 1018 CAS.
T. A. Newton, Y.-C. Huang, L. A. Lepak and M. A. Hines, J. Chem. Phys., 1999, 111, 9125 CrossRefCAS.
X.-Y. Zhu, J. A. Boiadjiev, J. A. Mulder, R. P. Hsung and R. C. Major, Langmuir, 2000, 16, 6766 CrossRefCAS.
K. Fujimori, in Organic Peroxides, ed. W. Ando, John Wiley & Sons, New York, 1992, p. 318 Search PubMed.
N. Y. Kim and P. E. Laibinis, J. Am. Chem. Soc., 1997, 119, 2297 CrossRefCAS.
N. Y. Kim and P. E. Laibinis, J. Am. Chem. Soc., 1998, 120, 4516 CrossRefCAS.
D. D. M. Wayner and A. Houmam, Acta Chem. Scand., 1998, 52, 377 Search PubMed.
R. A. Rossi, Aromatic Substitution by the SRN1 Mechanism, American Chemical Society, Washington, D.C., 1983 Search PubMed.
E. Southern, K. Mir and M. Shchepinov, Nature Genetics, 1999, 21, 5 Search PubMed.
F. Ozanam, C. Vieillard, M. Warntjes, T. Dubois, M. Paul and J.-N. Chazalviel, Can. J. Chem. Eng., 1998, 76, 1020 Search PubMed.
W. Feng and B. Miller, Langmuir, 1999, 15, 3152 CrossRefCAS.
R. Boukherroub and D. D. M. Wayner, J. Am. Chem. Soc., 1999, 121, 11513 CrossRefCAS.
P. Wagner, S. Nock, J. A. Spudich, W. D. Volkmuth, S. Chu, R. L. Cicero, C. P. Wade, M. R. Linford and C. E. D. Chidsey, J. Struct. Biol., 1997, 119, 189 CrossRefCAS.
R. L. Cicero, P. Wagner, M. R. Linford, C. J. Hawker, R. M. Waymouth and C. E. D. Chidsey, Polym. Prepr.(Am. Chem. Soc., Div. Polym. Chem.), 1997, 38, 904 Search PubMed.
T. Strother, W. Cai, X. Zhao, R. J. Hamers and L. M. Smith, J. Am. Chem. Soc., 2000, 122, 1205 CrossRefCAS.
T. Strother, R. J. Hamers and L. M. Smith, Nucleic Acids Res., 2000, 28, 3535 CrossRefCAS.
W. J. Royea, D. J. Michalak and N. S. Lewis, Appl. Phys. Lett., 2000, 77, 1988 CrossRefCAS.
A. Huang, O. R. Scherman, R. H. Grubbs and N. S. Lewis, Langmuir, 2001, 17, 1321 CrossRef.

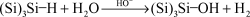
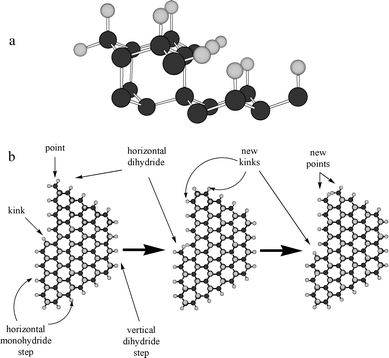
![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) direction and a vertical dihydride in the (
) direction and a vertical dihydride in the (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)



![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Me
CHCO2Me