High-resolution electron momentum spectroscopy of molecules
Michael J.
Brunger
* and
William
Adcock
School of Chemistry, Physics and Earth Sciences, The Flinders University of South Australia, G.P.O. Box 2100, Adelaide, S.A. 5001, Australia
First published on 22nd November 2001
Abstract
Covering: 1996–2001
1 Introduction
Electron momentum spectroscopy (EMS), or (e,2e) coincidence spectroscopy, is a unique tool for measuring the orbital momentum distribution for binding-energy selected electrons in atoms and molecules. Hence, it is an experimental technique intimately concerned with the details of the AOs or MOs which make up a given atom or molecule. Consequently, EMS is a potentially valuable means by which the chemists' orbital picture of molecules can be further refined. Photoelectron spectroscopy (PES) has played an important role in this regard1 by permitting very accurate orbital energies to be measured and, in conjunction with molecular orbital calculations, their sequence determined. As a result much progress has been made in the understanding of orbital interactions2 (e.g. through-space and through-bond) which govern the physical and chemical properties of molecules. EMS can be viewed as a complementary tool which is able to provide crucial details not attainable by PES, namely, orbital electron densities or momentum distributions. Because the technique is highly sensitive to the low-momentum region it is a particularly insightful probe of the chemically reactive outer spatial regions of the orbital electron momentum distribution of molecules. As a consequence of this, the application of EMS to atomic and molecular structure studies is expanding quite rapidly, as demonstrated by the fact that it is currently being carried out in at least eleven centres in different parts of the world.3The history and development of low-resolution† EMS has been well documented in a number of quite recent general reviews4–8 and the specific articles of McCarthy.9,10 Hence we do not repeat that detail again here except to note that since the pioneering study on methane,11 there have been about 100 molecules investigated8 using low-resolution EMS. The purpose of this article is, however, to consider high-resolution electron momentum spectroscopy (HREMS), its application with density functional theory (DFT) to some exemplar molecules, and, as a part of this application, to review how HREMS can elucidate the chemically interesting properties of those molecules.
High-resolution electron momentum spectroscopy refers to experiments conducted with an overall coincidence energy resolution of 0.6 eV (FWHM), or better. While we agree this figure is somewhat arbitrary, it simply reflects a value that is typically a factor of 2–3 times less than that obtained in most of the low-resolution EMS studies. HREMS of atoms and molecules originated at Flinders University in 1997 with the commissioning of their (e,2e) monochromator.12,13 Since that time it has been applied to several atoms14 and molecules,15–18 with its particular importance being that it has enabled conventional EMS to be extended to larger molecules, which are often the species where questions pertaining to the nature of their chemical bonding remain open.
In the next section of this review we briefly describe some of the fundamental experimental aspects of HREMS. In section 3 the theoretical basis of EMS is outlined, as is the application of DFT to EMS. The chemical significance of conventional EMS and HREMS is described in section 4, while the application of HREMS to specific exemplar molecules is discussed in detail in section 5. Finally, in section 6, some conclusions are drawn and future prospects for HREMS are considered.
2 Experimental details
The EMS technique and its theoretical analysis have been discussed in detail elsewhere.4 Briefly, however, EMS and HREMS are based on kinematically complete ionisation experiments (see Fig. 1) which are initiated by electrons of energy so high that the target structure is determined independently of their incident energy. For molecules, which are the gaseous targets of relevance to this review, this is in the region of 1 keV or above. Note that the target is considered to be at rest, as its thermal energy and momentum can be neglected for the process under consideration.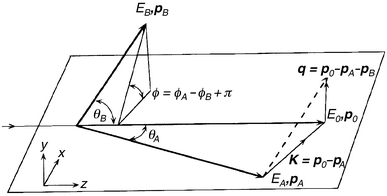 | ||
| Fig. 1 Schematic diagram illustrating the kinematics of an (e,2e) collision. The subscripts 0, A and B refer to the incident, scattered and ejected electrons, while p and E respectively denote their momenta and energy. q is the ion recoil momentum and K the momentum transfer. The plane in the figure denotes the scattering plane. θA and θB are the in-plane polar angles for the scattered and ejected electrons, while φA and φB are the out-of-plane azimuthal angles for the scattered and ejected electrons. | ||
We can write the ionising collision process as shown in eqn (1)
| e0(p0,E0) + T → T+f + eA(pA,EA) + eB(pB,EB) | (1) |
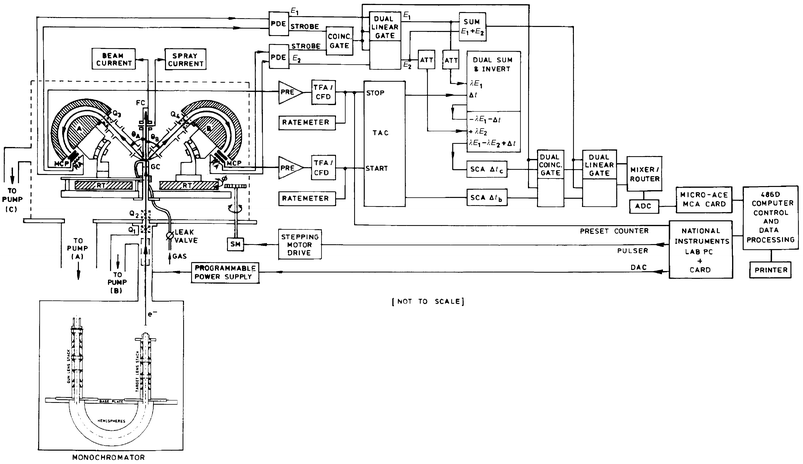 | ||
| Fig. 2 Schematic diagram of the Flinders symmetric noncoplanar HREMS spectrometer. | ||
In the studies we consider later in this review, noncoplanar symmetric kinematics is employed, with the two outgoing electrons, denoted by A and B, having essentially equal energies (500 eV for [1.1.1]propellane and cubane and 750 eV for allene and norbornadiene) and making equal polar angles (θ = 45°) with respect to the incident electron beam (see Fig. 1). The incident electron energy is E0 (1000 eV for [1.1.1]propellane and cubane and 1500 eV for allene and norbornadiene) plus the binding energy εf of the struck electron (eqn. (2)).
| E 0 = EA + EB + εf | (2) |
Eqn. (2) is simply a consequence of energy having to be conserved in the kinematics of the reaction.19
The ion recoil momentum q (and thus the momentum p of the target electron) is determined from momentum having to be conserved during the collision process (eqn. (3)).
| q = p0 − pA − pB | (3) |
At high enough energies and momentum transfer |p0 − pA|, momentum is transferred to the outgoing electrons only by a collision of the incident electron with a moving target electron of momentum p. In this case, eqn. (4) can be derived.19
| p = −q | (4) |
The complete valence region of the atom or molecule of interest is typically studied in several experimental runs, to check for self-consistency, using the Flinders symmetric noncoplanar EMS spectrometer4,19 (see Fig. 2). Both electron energy analysers have position sensitive detectors in their energy-dispersing planes. A full description of the coincidence spectrometer and the method of taking data can be found in McCarthy and Weigold,4 although we note that since their report there have been three major developments to this apparatus. Specifically, the computer hardware and operating system have been upgraded, the collision region is now differentially pumped, and, finally, an electron monochromator has been brought on-line. A full discussion of these developments can be found elsewhere8 although we note that the (e,2e) monochromator is designed around electrostatic lenses of cylindrical symmetry and incorporates a hemispherical selector.12,13 The lenses and apertures of the monochromator transport and collimate the electron beam, produced through thermionic emission from a tungsten hairpin filament, and ultimately image it, after energy monochromation, and with the help of a further einzel lens,20 onto the interaction region. The (e,2e) monochromator typically produces electron beam currents at the interaction or collision region in the range 20–40 μA, with a percentage focus into the Faraday cups always greater than 98% during operation. The lower beam currents obtained using the (e,2e) monochromator, compared to low-resolution EMS studies (50–100 μA), would normally lead to lower (e,2e) count rates. However this effect is at least partly offset at Flinders with the incorporation of the aforementioned differential pumping of the collision region, which allows us to operate at higher target beam densities.
In the experiments we consider later in section 5, the typical binding energy range of interest (εf = 3.5–46.5 eV) is stepped through sequentially at each of a chosen set of ϕ angles (ϕ = 0–30°) using a binning mode19 through the entire set of azimuthal angles ϕ. Note that the term “binning mode” refers to an experimental procedure by which we ensure that any non-uniform response in the MCP/RA particle detectors is averaged out during the course of an experiment. This further ensures that our measured binding-energy spectra are free from any possible unwanted instrumental effects. Scanning through a range of ϕ is equivalent to sampling different target electron momenta as, for the noncoplanar symmetric geometry of our experiments, p is given by eqn. (5).
| p = [(2pAcos θ − p0)2 + 4pA2sin2 θsin2 (φ /2)]1 /2 | (5) |
Note that the transform p ↔ ϕ, as embodied in eqn. (5) above, follows from the conservation of momentum (eqn. (3)), the binary encounter approximation8 (eqn. (4)) and from the appropriate vector addition analysis in Fig. 1 with EA = EB, θA = θB = θ(= 45°), φA = 0 and φ = π − φB set for our noncoplanar symmetric geometry. As mentioned previously, ϕ is varied in practice by rotating the ejected-electron analyser B into and out from the scattering plane. It then follows directly from eqn. (5) that by varying ϕ we vary the target electron momentum p.
Using eqn. (5), and also allowing for angular resolution effects,21 then given the above range of ϕ, the target electron momenta usually vary from about 0.07 to about 2.5 ℏ/a0 (atomic units) in the Flinders experiments.
The energy resolution (ΔEres) of the present HREMS studies, as determined from measurements of the binding energy spectra of helium, is in the range 0.5–0.6 eV (FWHM). The exception to this was the experiment with [1.1.1]propellane, which was performed at a somewhat lower resolution. The angular resolution was about Δϕ = 1.2°, Δθ = 0.6°, as determined from the analyser electron optics and apertures and from a consideration of the argon 3p angular correlation.
The analysis of the measured binding energy spectra, at each ϕ, has also been described many times before (see, for example, Adcock et al.15). Briefly, a least squares fit to the spectra,22 assuming Gaussian profiles, is performed. The binding energies of the respective orbitals are usually fixed at the known photoelectron spectroscopy (PES) values, although satellite lines can complicate this, and the Gaussian widths are a convolution of the energy resolution and the natural line widths of the respective orbitals. The area under each profile, and its uncertainty, are determined in the fit at each value of ϕ. This process thus allows us to derive the required momentum distributions (area as a function of ϕ) for the respective valence orbitals of the molecule under study. Although the measured momentum distributions (MDs) are not absolute, relative magnitudes for the different transitions are obtained.4 In all the HREMS studies we have conducted, the experimental MDs are placed on an absolute scale by summing the experimental flux for each measured ϕ for all the relevant outer valence orbitals (of a given molecule), and then normalising this to the corresponding sum from the result of one of our plane-wave impulse approximation (PWIA) calculations (for that same molecule). A full discussion of the PWIA calculations is given in the next section.
Since conventional EMS and HREMS are both very sensitive to impurities, great care needs to be exercised to minimise the possibility of sample contamination both during synthesis (e.g. [1.1.1]propellane and cubane) and in transportation from the storage reservoir to the interaction region. The allene and norbornadiene sources were commercially purchased with high purity. Note that the sample gas driving pressure was too low to cause any significant clustering by supersonic expansion. The results of each scan are carefully monitored for any signs of sample degradation and samples are regularly changed to additionally minimise the possibility of sample contamination or degradation.
3 Theoretical basis of EMS and its application with DFT
In the independent-particle (e.g. Hartree–Fock or density-functional) model each ion state corresponds to a molecular orbital. Normalisation of momentum density gives the orbital occupation number. Each independent-particle ion state is split into a symmetry manifold by electron correlation with each state (satellite) of a symmetry manifold being uniquely identified by the shape of its MD. The summed momentum density for the manifold is equal to the momentum density of the corresponding orbital. The fraction of the orbital manifold density belonging to an ion state is the spectroscopic factor (which sum to unity for each manifold).The PWIA is used to analyse the measured cross sections for high-momentum transfer (e,2e) collisions.19 If ionisation can be considered to proceed by a direct collision between the incident electron and the target electron, with all of the momentum lost by the incident electron transferred to the ejected electron, we have the so-called binary regime. If, to go one step further, it can also be assumed the incident electron only interacts with the ejected electron and neither affects the target nor is affected by the target, we have the impulse approximation. Under these circumstances the wave function for the target is the isolated-target wave function, and the final-residual-ion wave function is the isolated wave function of the ion. Combining plane waves with the impulse approximation gives the PWIA, where the incident, scattered and ejected electrons are all taken to be plane waves.8 We expect that this approximation will be a good one when the energies of all the interactions are small compared to the kinetic energies of the incident, scattered and ejected electrons. In practice, this means that the binding energy of the ejected electron is small compared to the kinetic energies of the incident and outgoing electrons. The PWIA is the approximation most often used in extracting momentum distribution information from the (e,2e) cross sections, as we explicitly see below.
Using the Born–Oppenheimer approximation for the target and ion wave functions, the (e,2e) differential cross section σ, for randomly oriented molecules and unresolved rotational and vibrational states, is given by eqn. (6) where K is a kinematic factor which is essentially constant in the present experimental arrangement, ΨN − 1f and ΨNi are the electronic many-body wave functions for the final ((N − 1) electron ion) and target (N electron) ground states, and p is the momentum of the bound electron at the instant of ionisation.
| σ = K∫dΩ|〈pΨN − 1f|ΨNi〉|2 | (6) |
The ∫dΩ term denotes an integral over all angles (spherical averaging) due to averaging over all initial rotational states. The average over the initial vibrational states is well approximated by evaluating orbitals at the equilibrium geometry of the molecule. Final rotational and vibrational states are eliminated by closure.
The momentum-space target-ion overlap23 〈pΨN − 1f|ΨNi〉 can be evaluated using configuration interaction (CI) descriptions of the many-body wave functions, but usually the weak-coupling approximation4 is made. Here the target-ion overlap is replaced by the relevant orbital of, for example, the Hartree–Fock or Kohn–Sham24 ground state Φi, multiplied by a spectroscopic amplitude, which is the coefficient, in the CI description of the ion state, of the configuration representing a hole in the appropriate ground state orbital. With these approximations eqn. (6) reduces to eqn. (7) where ϕj(p) is the momentum space orbital.
| σ = KS(f)j∫dΩ|ϕj(p)|2 | (7) |
The spectroscopic factor S(f)j is the square of the spectroscopic amplitude for orbital j and ion state f. It satisfies the sum rule (eqn. (8)).
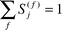
| (8) |
Hence it may be considered as the probability of finding the one-hole configuration in the many-body wave function of the ion.
The target-ion overlap is a one-electron function called the quasi-particle orbital. A quasi-particle equation, the Dyson equation, can be constructed from the electronic Schrödinger equations for the target and ion.23 Formally this is a one-electron Schrödinger equation with target and ion structure details contained in the potential operator. The quasi-particle energies are given by the poles in the Green's function of this equation, which can be evaluated using diagrammatic perturbation theory.25
The Kohn–Sham equation24 of DFT may be considered as an approximate quasi-particle equation, with the potential operator approximated by the exchange-correlation (XC) potential.23 Usually this is done at the local spin density (LSD) approximation level (sometimes also known as the local density approximation (LDA)), although in parts of our work we also employ non-local correlation functional corrections. Duffy et al.26 showed the physical significance of the valence orbitals of DFT by demonstrating their ability to describe EMS data that are not well described by SCF calculations that omit electron-correlation considerations, but are well described by full CI calculations.
In order to compute the coordinate space Kohn–Sham orbitals ψj we employed DGauss, a program package developed at CRAY Research by Andzelm and colleagues.27,28 DGauss is itself part of UniChem, a suite of computational quantum-chemistry programs from Oxford Molecular (Pharmacopeia). UniChem was used to build the molecular structures (allene, [1.1.1]propellane, cubane and norbornadiene) then DGauss employed to minimise the energies by optimising the geometries. The molecular coordinates at the optimum geometry (minimum energy) and the Gaussian molecular orbital parameters (coefficients and exponents) were next treated as input to the Flinders-developed AMOLD programme,4 which computes the momentum space spherically averaged molecular-structure factor19 and the (e,2e) cross section or MD.
The comparisons of calculated momentum profiles with experiment (see section 5) may be viewed as an exceptionally detailed test of the quality of the basis set. In the studies we describe later in this review, up to seven basis sets have been employed in the DFT computations. These basis sets are denoted by the acronyms DZ94, DZ94P, DZVP, DZVP2, TZ94, TZ94P and TZVP. The notations DZ and TZ denote basis sets of double- or triple-ζ quality. V denotes a calculation in which such a basis is used only for the valence orbitals and a minimal basis is used for the less chemically reactive core orbitals. The inclusion of long-range polarisation functions is denoted by P. The DZ94, DZ94P, DZVP, DZVP2, TZ94, TZ94P and TZVP basis sets are specially designed for DFT calculations27,29 giving the respective contraction schemes of (621/41), (621/41/1), (621/41/1), (721/1/1), (7111/411), (7111/411/1) and (7111/411/1) for carbon and (41), (41), (41), (41/1), (311), (311/1) and (3111/1) for hydrogen. Note that this notation also indicates the number of primitive Gaussians. For example, (621/41/1) means there are 3 contracted s, 2 contracted p and 1 contracted d functions. The s functions consist of 6, 2 and 1 primitive Gaussians while the p functions consist of 4 and 1 primitive Gaussians. Corresponding to these orbital basis sets are auxiliary basis sets to represent the electron density, the XC potential and energy. The auxiliary basis set corresponding to the DZ94, DZ94P, DZVP, DZVP2, TZ94, TZ94P and TZVP orbital basis sets is called A2,30 in which the s-, p- and d-orbital exponentials were determined separately from an optimisation that reproduces as accurately as possible the energy from an atomic DFT calculation. The contraction schemes of the A2 basis sets for H and C are (4/1) and (8/4/4), respectively.
The DFT calculations were performed using both the LSD (LDA) and generalised gradient approximation (GGA) methods. The GGA methods use various gradient-corrected functionals based on the Becke–Perdew (BP) XC functionals—Becke31 for exchange (X) and Perdew32,33 for correlation (C) in the calculations. Other XC functionals such as the Becke–Wang–Perdew (BWP34,35), Becke–Lee–Yang–Parr (BLYP35) and Wang–Perdew (WP) are also employed in the geometry optimisations. The LSD method invokes an LSD approximation using the Dirac exchange energy functional and the Vosko–Wilk–Nusair (VWN36) local spin density approximation level correlation energy functional. The calculations were performed on an SGI-R500-02 work station and a CRAY J90se/82048 computer employing the computer distribution technique. Note that the term computer distribution technique is simply a shorthand notation to denote that the calculations were set up on the SGI-2 work station before being launched on the CRAY supercomputer. In addition, for the [1.1.1]propellane molecule, specifically to allow for comparison with the results from the DFT orbitals, we also employed SCF orbitals computed by GAMESS37 using Dunning38 basis sets at the triple-ζ-plus polarisation level. This SCF calculation used the optimised structural geometry of Wiberg.39
Some of the DFT results we obtained,17 for the ground electronic state of allene, with the DZVP, DZVP2 and TZVP basis sets and XC functionals are summarised in Tables 1 and 2. It is clear from these tables that the calculated electronic structure and the calculated harmonic vibrational frequencies are rather sensitive to the basis set and XC functional used in the computation. Thus a technique which a priori provides an indication for the most physically reasonable representation of the molecule, allene in this particular case, would be invaluable for determining which of the calculations was the most accurate. This point is explored further in the following sections.
| Method | Basis set | r CC/Å | r CH/Å | ∠HCC/° | E/Eh |
|---|---|---|---|---|---|
| GGA-BP | TZVP | 1.311 | 1.094 | 120.962 | −116.701196 |
| GGA-BP | DZVP2 | 1.321 | 1.096 | 120.612 | −116.684742 |
| GGA-BWP | TZVP | 1.311 | 1.094 | 120.962 | −116.681426 |
| GGA-BLYP | TZVP | 1.312 | 1.092 | 121.015 | −116.647641 |
| GGA-WP | TZVP | 1.309 | 1.092 | 121.012 | −116.642647 |
| LSD-VWN | TZVP | 1.302 | 1.098 | 120.028 | −115.594469 |
| HF-SCF40 | STO-3G40 | 1.308 | 1.082 | 120.000 | −115.900785 |
| Method | Basis set | ν 1 = ν2/cm−1 | Int./km mol−1 | ν 3 = ν4/cm−1 | Int./km mol−1 | ν 9 /cm−1 | Int./km mol−1 | ν 11 /cm−1 | Int./km mol−1 |
|---|---|---|---|---|---|---|---|---|---|
| a Zero-point vibrational energy is ≊33.4 kcal mol−1. | |||||||||
| GGA-BP | DZVP | 372.493 | 9.43 | 769.529 | 59.56 | 1363.362 | 7.68 | 1998.840 | 87.48 |
| GGA-BP | DZVP2 | 355.563 | 9.51 | 758.297 | 75.30 | 1364.290 | 8.98 | 1994.665 | 82.51 |
| GGA-BP | TZVP | 355.857 | 8.10 | 763.914 | 67.38 | 1374.987 | 6.51 | 1996.200 | 76.04 |
| GGA-BWP | TZVP | 374.635 | 9.05 | 775.795 | 59.81 | 1368.177 | 7.63 | 2000.022 | 85.52 |
| GGA-BLYP | TZVP | 368.465 | 8.63 | 775.420 | 57.49 | 1377.424 | 6.27 | 1985.142 | 83.12 |
| GGA-WP | TZVP | 372.440 | 9.29 | 774.253 | 61.85 | 1363.370 | 8.90 | 2007.746 | 87.30 |
| LSD-VWN | TZVP | 387.600 | 11.36 | 774.750 | 65.75 | 1327.010 | 15.43 | 2034.130 | 104.32 |
4 Chemical significance
Earlier articles that provide excellent descriptions for various aspects of the chemical applications and chemical significance of EMS include those from Winkler et al.,40 Brion,41 Neudatchin et al.,7 Coplan et al.,6 McCarthy and Weigold,4 Michalewicz et al.,42 Neville et al.,43 and Adcock et al.44Traditional quantum chemistry has been largely based on the position-space representation.6 This is intuitively reasonable as most of our chemical understanding is based on position-space pictures. Further, the Schrödinger equation in position-space is a differential equation, and considerable theoretical technology exists for solving differential equations. In momentum-space the Schrödinger equation is an integral equation, which is generally thought of as being more difficult to solve.8 For molecules, of course, the position representation is a multi-centred problem, whereas the momentum representation is a single-centre problem which in principle is simpler.4
Despite the obvious preference towards working in position-space, momentum-space chemistry, in fact, has a long history. Coulson and Duncanson,45–48 examined the electronic structure of molecules and bonding concepts in momentum-space. In these papers they clearly put forward a case for the utility of momentum-space work offering new insights into molecular structure. Their work was later extended by, for example, Epstein49 and Epstein and Tanner50 when Compton scattering became a practical tool for investigating the structure of molecules. The ability of the EMS (or HREMS) technique to select individual final states (orbitals) and to measure the associated spherically averaged MDs, prompted a renewed interest in the understanding of chemical bonding directly in momentum-space.
Momentum-space chemistry has, more recently, also been employed to study molecular similarity and dissimilarity for biomolecules, bond formation and hyperpolarisabilities of a range of molecules.51,52 The momentum-space approach is particularly suited to problems for which the molecular activity depends less on the details of the bonding topology than on features of the long-range slowly varying valence charge density. This facet was utilised successfully by McCoy and Sykes53 in their work on estimating the molecular properties of a wide range of molecular species using momentum-space wave functions.
The binding-energy spectra for the complete valence region of molecules generally exhibits strong final-state correlations in the inner valence region. The MDs for transitions to different final states reflect the orbital symmetries and permit allocation of each transition (satellite) to a particular manifold. This leads to clarification in orbital ordering as well as to the role of electron–electron correlations in the electronic structure of the molecule and its ion.
The independent-particle orbital approximation has generally been applied to partition the total wave function Ψ into orbital wave functions φi. The Hartree–Fock (HF) canonical orbitals obtained in self-consistent-field (SCF) procedures are typically used.8 In the SCF calculations energy minimisation, consistent with the procedures of the variational theorem, is employed as a criterion for determining how reasonable the physical representation of a wave function is. Such energy-optimised wave functions are usually quite good near the “inner” regions of the molecule and for calculating energy-related properties of molecules. This is because the strong-potential regions, i.e. high-momentum regions, near the nuclei account for most of the energy. They are not, however, necessarily accurate in the “outer” (low momentum) region of the molecule, where the wave function determines properties such as the dipole and quadrupole moments. It is the outer region of molecules that is of prime importance when determining chemical reactivity and bonding, as well as in physical phenomena involving intermolecular forces, such as hydrogen bonding and van der Waals interactions.54 EMS has the advantage that it is most sensitive to the low-momentum region of the wave function, which mainly corresponds to the outer region in coordinate space. For orbitals that populate the outer region, EMS has shown that the DFT method, which optimises the density rather than the energy, gives a better description of the MD than the HF method.26,55 DFT orbitals also give an excellent account of other measurable properties of molecules such as molecular structure, electric dipole moments, vibrational spectra and NMR.55
In summary, we believe conventional and high-resolution EMS is of considerable interest to chemists for three principal reasons.
(i) EMS and HREMS offers the intriguing possibility of imaging molecular orbitals. As Coplan et al.6 state “…MDs are only a phase factor and Fourier transform away from the wave function from which all observable properties of atoms and molecules can, in principle, be obtained”.
(ii) Within the PWIA and, in many cases, the target-Hartree–Fock-approximation (THFA), the measured (e,2e) cross section (MD) may be directly compared with the calculated spherically averaged MD of a specific molecular orbital, once the appropriate angular-resolution function has been folded in.21 Hence EMS is also a powerful technique for evaluating the a priori quality of theoretical wave functions in quantum chemistry.5,56
(iii) It is of considerable interest to use EMS and HREMS to study properties of molecules with unusual characteristics such as strained geometries and unusual hybridisations or electronic properties. EMS or HREMS coupled with DFT calculations may elucidate significant molecular features difficult to study by other techniques.
Chemical considerations influenced the choice of the four exemplar molecules we examine in the next section: allene, [1.1.1]propellane, cubane and norbornadiene (see Figs 3a–d respectively). Propellane and cubane, containing unusual bonding or atom hybridisation, are likely to provide a very stringent test of basis set quality. Allene is an important organic reagent whose electronic structure has been topical, at least in part, due to its unique ground-state molecular geometry.57,58 Finally, the electronic structure of norbornadiene is a prototype for the study of through-space and through-bond interactions, concepts originally introduced by Hoffmann et al.59,60 and incorporated into an SCF scheme by Heilbronner and Schmelzer.61
![Structural representations of (a) allene, (b) [1.1.1]propellane, (c) cubane and (d) norbornadiene.](/image/article/2002/P2/b100700i/b100700i-f3.gif) | ||
| Fig. 3 Structural representations of (a) allene, (b) [1.1.1]propellane, (c) cubane and (d) norbornadiene. | ||
5 Exemplar molecules
5.1 Allene (C3H4)
Allene or propa-1,2-diene has the structure H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH2 and as such it is the simplest hydrocarbon to contain two double bonds (see Fig. 3a). Braidwood et al.62 undertook the only low-resolution EMS study into its properties, although PES interest is highlighted by the work of Baker and Turner,63 Thomas and Thompson,64 Bieri et al.,65 Yang et al.66 and Baltzer et al.67
CCH2 and as such it is the simplest hydrocarbon to contain two double bonds (see Fig. 3a). Braidwood et al.62 undertook the only low-resolution EMS study into its properties, although PES interest is highlighted by the work of Baker and Turner,63 Thomas and Thompson,64 Bieri et al.,65 Yang et al.66 and Baltzer et al.67
Braidwood et al.62 measured binding-energy spectra of allene using their low-resolution noncoplanar symmetric electron-coincidence spectrometer. From such spectra (see section 2) they derived experimental MDs for allene's 2e, 1e + 2b2, 2a1, 1b2 and 1a1 valence molecular orbitals (MOs). Theoretical MDs were also calculated by Braidwood et al. using a PWIA description for the ionisation mechanism4 and a HF wave function constructed from the GAMESS program.37 Braidwood et al. found only marginal agreement between their measured and calculated MDs. One possible reason for this discrepancy was that their STO-3G basis was an inadequate representation for the MOs. Another possible explanation might be due to problems with their62 binding-energy spectra measurements or in the spectral deconvolution procedure employed by them to derive their MDs. Indeed Braidwood et al. noted some ambiguities with their work. For instance, they observed a “feature” in their binding energy spectra, centred at εf = 20 eV, which is not seen in any of the relevant PES studies.65,67 Such a “peak”, which they had to include in their analysis to account for the measured coincidence signal in their binding-energy spectra, could arise due to either the low energy resolution employed by Braidwood et al.,62 making the unique spectral deconvolution of overlapping features problematic, and/or as a result of employing Gaussian functions in their62 spectral deconvolution whereas the line profiles could in principle be quite asymmetric. Consequently Wang et al.17 performed new high-resolution (ΔEres ≈ 0.49 eV FWHM) EMS measurements, under almost identical kinematic conditions as Braidwood et al., in order to resolve some of these ambiguities and also to provide a stringent cross check for the previously reported MDs. In addition, Wang et al.17 also used their measurements to investigate the validity of the spectroscopic sum rule (see eqn. (8)) for allene, which was the first time this was attempted for molecules.
In light of the marginal agreement between experiment and theory for the MDs, as found by Braidwood et al.,62 Nicholson et al.68 employed a numerical inverse method of extracting the target-ion overlap, or normalised Dyson orbital. This was specifically done for the 2e highest-occupied-molecular-orbital (HOMO) by using a quantum-mechanically constrained fitting procedure69 and the PWIA to describe the ionisation mechanism. The result68 of employing the inverse method to the 2e experimental MD of Braidwood et al. is given in Fig. 4. Clearly Nicholson et al.'s estimate of the normalised Dyson orbital provided a much better fit to the experimental MD than did that obtained using the GAMESS basis 2e orbital alone (see Fig. 4). This was well reflected by the value of χ2 falling from 372, when the 2e HF STO-3G orbital was employed, to a value of 2.3 when the normalised Dyson orbital was used. As expected Nicholson et al.68 found the 2e basis orbital coefficient dominated the normalised Dyson orbital, the high-quality fit of Fig. 4 being achieved with only a 6% 1e and unoccupied orbital contribution. Nonetheless this 6% contribution is significant in another sense as it clearly indicated the importance of electron-correlation effects in the allene molecule. Note that electron correlation effects, such as final-state-configuration-interaction,4 do not invalidate our orbital-by-orbital MD analysis (see later). While correlation effects can potentially lead to a significant splitting of the manifold spectral strength into satellites, thereby complicating the interpretation of the measured binding-energy spectra, as noted previously the MD of each satellite retains the same form as that for the manifold as a whole. DFT, by its very construction, already allows for electron-correlation considerations in the calculation of the Kohn–Sham orbitals.24 Consequently, by using DFT basis sets Wang et al.17 thought they might determine PWIA-DFT MDs in better agreement with their experimental results and those of Braidwood et al.,62 than Braidwood et al. found earlier with their HF-level orbitals. Wang et al. therefore carried out DFT computations to generate the molecular information required by the PWIA to calculate MDs for the valence MOs of allene. Three sets of DFT basis sets at the TZVP, DZVP and DZVP2 levels were employed in that study.17 These calculations were carried out under the LSD approximation, both with and without non-local corrections.
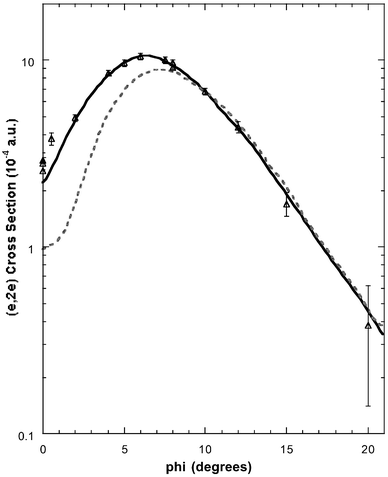 | ||
| Fig. 4 The 1500 eV symmetric noncoplanar MD for the 2e HOMO of allene. The data of Braidwood et al.62 (△) are compared against the PWIA-SCF results of Braidwood et al.62 (- - - - -) and the PWIA–normalised Dyson orbital result of Nicholson et al.68 (——). Reproduced with permission of John Wiley and Sons. | ||
The final significant aspect of their application of HREMS to allene was that by comparing the experimental and theoretical MDs, for the 2e, 1e + 2b2, 2a1, 1b2 and 1a1 valence MOs, Wang et al.17a priori independently determined which of the DFT basis sets they studied provided the most physically reasonable representation of the allene molecule. We have previously discussed15,16,40,70 that such a procedure is highly sensitive in its ability to differentiate between the quality of various basis sets and that once this is achieved one can then utilise this “optimum” wave function to extract the chemically important molecular property information for the system under study. This is precisely what Wang et al.17 did for allene, as we demonstrate shortly.
Typical binding-energy spectra of allene in the region 6–34.5 eV and at a total energy E = 1500 eV are given in Fig. 5. These spectra were subsequently analysed17 and the required MDs for all the respective valence orbitals of allene derived. Before sequentially examining the MDs for each of the valence MOs, we make some general observations as to the results embodied in Figs. 5–11. Firstly, the typical binding-energy spectra from Wang et al.17 (Fig. 5) are in excellent qualitative agreement with the corresponding PES results.65,67 There is no “feature”, centred at εf = 20 eV, observed in these HREMS binding-energy spectra, thereby resolving the ambiguity that had previously existed between the result of Braidwood et al.62 and the PES studies.65,67 Secondly, it is clear from Figs. 6–11 that the MDs of Wang et al.17 and the earlier MDs of Braidwood et al. are in good agreement with one another for each of the respective valence MOs and across all measured ϕ (or p). The MDs of Wang et al. therefore confirm the earlier results.62 Finally, the PWIA-DFT calculations of Wang et al. are all in better agreement, again for each respective valence MO, with their MD data and the MD data of Braidwood et al.62 than are the earlier PWIA-SCF (STO-3G) level calculations from Braidwood et al. This is specifically illustrated in Figs. 4 and 6 for the 2e HOMO. Hence the DFT calculations of Wang et al. provided a more physical representation for the allene molecule than did the earlier HF computation.62
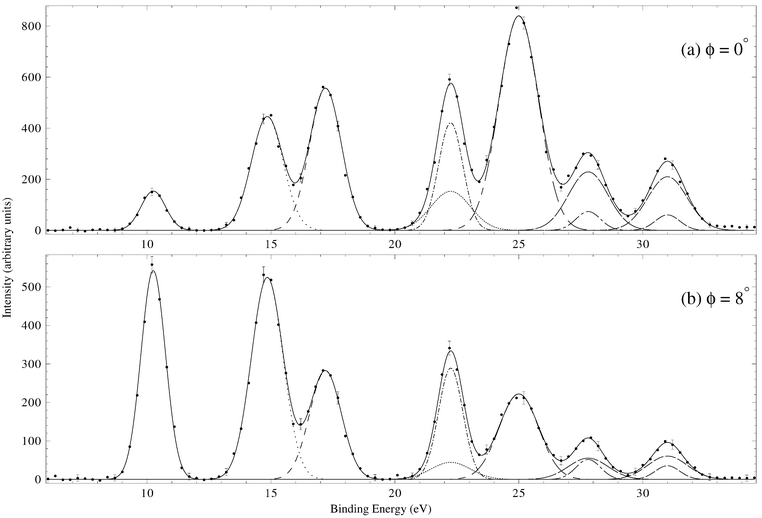 | ||
| Fig. 5 Typical binding-energy spectra from the 1500 eV noncoplanar symmetric HREMS investigation of Wang et al.17 into allene. The curves show the fits to the spectra at (a) ϕ = 0° (p ≈ 0.03 ℏ/a0) and (b) ϕ = 8° (p ≈ 0.73 ℏ/a0) using the known energy resolution. The solid line is the overall result from the fitting (deconvolution) process, the other lines (dotted, dashed etc.) represent the contribution of each Gaussian to the overall fit. Reproduced with permission of John Wiley and Sons. | ||
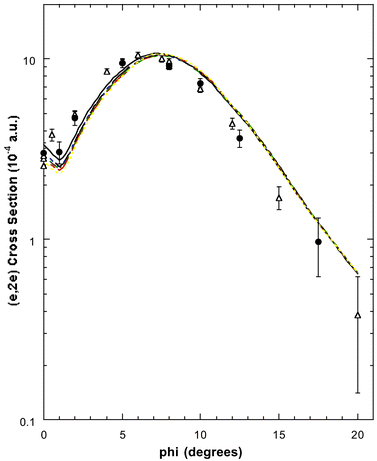 | ||
| Fig. 6 The 1500 eV symmetric noncoplanar MD for the 2e HOMO of allene. The data of Wang et al.17 (●) and the earlier data of Braidwood et al.62 (△) are compared against the results of the PWIA-DFT calculations:17 (——) BLYP/TZVP, (- ——) LSD/TZVP, (— —) BWP/TZVP, (- - - - -) WP/TZVP, (-- --) BP/TZVP, (· — · —) BP/DZVP2 and (·· ·· ··) BP/DZVP. Acronyms are defined in the text. Reproduced with permission of John Wiley and Sons. | ||
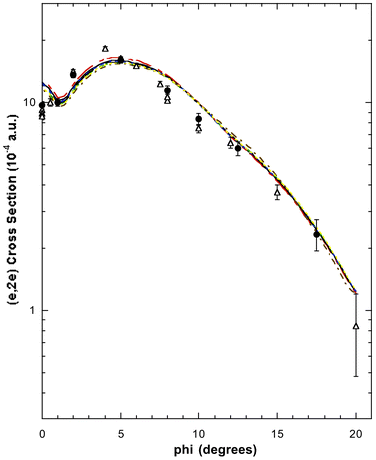 | ||
| Fig. 7 The 1500 eV symmetric noncoplanar MD for the 1e + 2b2 orbitals of allene. The legend is the same as that for Fig. 6. Reproduced with permission of John Wiley and Sons. | ||
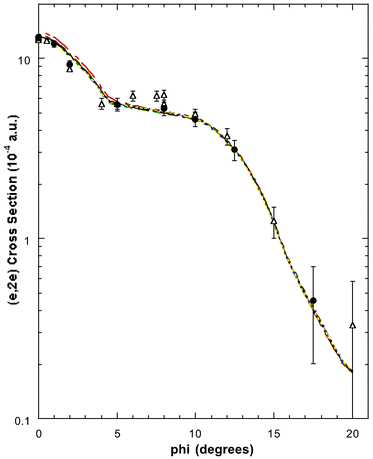 | ||
| Fig. 8 The 1500 eV symmetric noncoplanar MD for the 2a1 orbital of allene. The legend is the same as that for Fig. 6. Reproduced with permission of John Wiley and Sons. | ||
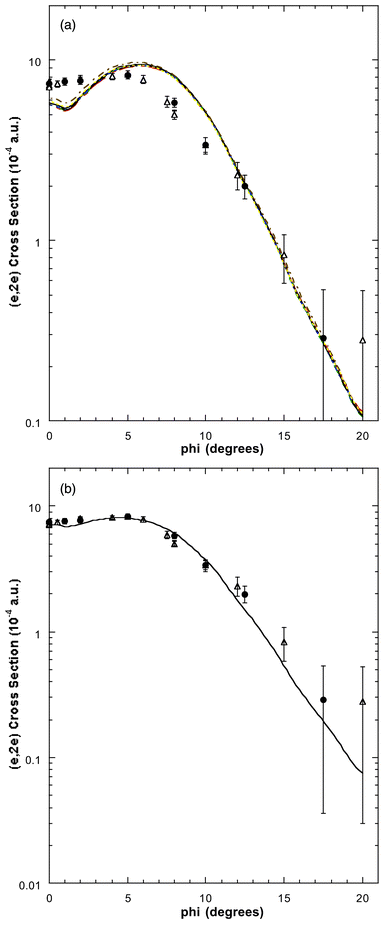 | ||
| Fig. 9 (a) The 1500 eV symmetric noncoplanar MD for the 1b2 orbital of allene. The legend is the same as that for Fig. 6. Reproduced with permission of John Wiley and Sons. (b) The 1500 eV symmetric noncoplanar MD for peak 4 (εf = 22.25 eV) of the binding-energy spectra. The data of Wang et al.17 (●) and Braidwood et al.62 (△) are compared against the result from the PWIA-DFT (BP/TZVP) calculation17 for 0.67 × 1b2 + 0.09 × 1a1 (——). Reproduced with permission of John Wiley and Sons. | ||
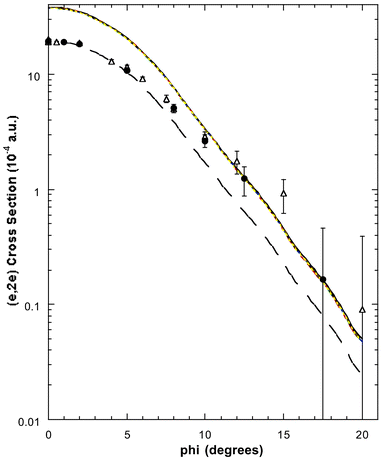 | ||
| Fig. 10 The 1500 eV symmetric noncoplanar MD for the 1a1 orbital of allene. The legend is the same as that for Fig. 6 except 0.51 × 1a1 (BP/TZVP) is additionally plotted (— —). Reproduced with permission of John Wiley and Sons. | ||
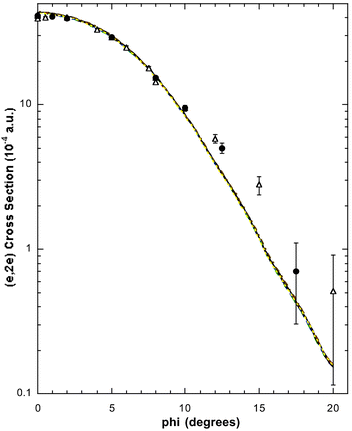 | ||
| Fig. 11 The 1500 eV symmetric noncoplanar MD for the 1b2 + 1a1 orbitals of allene. The legend is the same as that for Fig. 6. Note that a 10% continuum contribution (εf ≤ 33.5 eV), as determined by Braidwood et al.,62 is allowed for in the data of Wang et al.17 Reproduced with permission of John Wiley and Sons. | ||
In Fig. 6 we compare the experimental MD of Wang et al.17 and Braidwood et al.,62 for the 2e HOMO (εf = 10.25 eV), with the results from the PWIA-DFT computations for each of the three DFT basis states and five local/non-local XC functionals Wang et al. considered. It is apparent from Fig. 6 that all the PWIA-DFT computations are in fair overall agreement with the measured MDs, the theory somewhat underestimating the magnitude of the cross section for 1° < ϕ < 6° and somewhat overestimating the magnitude of the cross section for ϕ > 7.5°. For this orbital it was not easy to differentiate between the quality of the various DFT calculations, they all led to similar MDs (see Fig. 6) within a PWIA framework. However, on very close scrutiny, it was possible to discern that the TZVP basis with BLYP and BP XC functionals gave MDs in marginally better agreement with the experimental MD results, thus providing the best representation for the 2e orbital. All the PWIA-DFT calculations predicted a local minimum in the MD at ϕ ≊ 1°. The experimental MD results17,62 are not inconsistent with this theoretical prediction, which we believe originated from electron correlation effects. The comparison between the PWIA-DFT and experimental MDs provided strong evidence for all the 2e spectroscopic strength being located at εf = 10.25 eV. In other words the HREMS and EMS measurements predict a spectroscopic factor S2e ≈ 1 at εf = 10.25 eV. This observation was in excellent agreement with the results of the available many-body Green's function calculations.62,65,67
PES measurements63–65,67 predict the 1e and 2b2 MOs to be essentially degenerate with ionisation potential ∼15 eV. Consequently they were represented in the HREMS spectral deconvolution procedure by a single Gaussian (under peak 2 of the binding-energy spectra) at εf = 14.85 eV. The experimental 1e + 2b2 MD of Wang et al.,17 the 1e + 2b2 MD of Braidwood et al.62 and the PWIA-DFT 1e + 2b2 MD calculation results17 are all plotted in Fig. 7. Once again the overall level of agreement between the theory and experimental MDs is fair and, like that described above for the 2e orbital, all the DFT basis sets lead to theoretical (e,2e) cross sections that are very similar. The only exception to this general comment would appear to have been the PWIA-DFT (BP/DZVP) results for 10° < ϕ < 15°, where it tended to overestimate the magnitude of the cross section. The level of comparison between the experimental and theoretical MDs indicated that almost all of the 1e and 2b2 spectroscopic fluxes were identified under peak 2 of the binding-energy spectra. Thus the HREMS spectroscopic factors for the 1e and 2b2 orbitals at εf = 14.85 eV are both approximately one, i.e.S1e ≈ 1 and S2b2 ≈ 1 at εf = 14.85 eV. These results are in very good agreement with the predictions of the many-body Green's function calculations for the respective 1e and 2b2 orbitals.62,65,67
In Fig. 8 the HREMS 2a1 molecular orbital, at εf = 17.2 eV, theoretical and experimental MDs from Wang et al. are presented. Also shown in this figure are the MD measurements of Braidwood et al. With the exception of the PWIA-DFT (LSD/TZVP and BWP/TZVP) MDs, which overestimated the magnitude of the (e,2e) cross section at ϕ < 5°, all their17 DFT calculations were in good agreement with the experimental MD data. Here it appears that most, if not all, of the 2a1 spectral strength is encompassed under peak 3 at εf = 17.2 eV. Thus the HREMS spectroscopic factor for the 2a1 orbital at εf = 17.2 eV is S2a1 ≈ 1. Once again this result is in good accord with those calculated using many-body Green's function theories.62,65,67
The allene binding-energy spectra, consistent with many-body theory predictions, indicate strong final-state-configuration-interaction effects for the inner valence orbitals, thereby leading to a splitting and mixing of the 1b2 and 1a1 spectroscopic strength. As a consequence of this none of the Wang et al.17 1b2 PWIA-DFT MD calculation results are in agreement with their experimental peak 4 MD or the corresponding MD of Braidwood et al., for ϕ < 12°. In particular, the theory MDs underestimate the strength of the (e,2e) cross section for ϕ < 4° and overestimate its magnitude for 5° ≤ ϕ < 12°. This is clearly seen in Fig. 9a. On the other hand if we allow that under peak 4 of the binding-energy spectra, at εf = 22.25 eV, there is an admixture of 67% of the total 1b2 spectral strength and 9% of the total 1a1 spectral strength, the level of agreement, except for the DZVP2/BP basis and XC functional, between the PWIA-DFT MD calculations and the measured MDs is very good. This is specifically illustrated in Fig. 9b for the TZVP basis set and non-local BP-XC functional. Consequently, we can assign the spectroscopic factor of the 1b2 MO at εf = 22.25 eV to be S1b2 ≈ 0.67, while the spectroscopic factor of the 1a1 MO at εf = 22.25 eV is S1a1 ≈ 0.09.
Somewhat surprisingly, the spectral deconvolution of Wang et al.17 indicated only a very small 1b2 contribution (<2%) under peak 5 of the binding-energy spectra. Thus, most of the spectral strength under peak 5, at εf = 25.0 eV, must originate from the 1a1 orbital. In Fig. 10 we therefore plot their experimental17 1a1 MD, the 1a1 MD from Braidwood et al. and the result from Wang et al.'s PWIA-DFT calculations. As the inner valence 1a1 orbital is severely split by final-state-configuration-interaction effects,62,65,67 we anticipated the theoretical 1a1 result to overestimate the magnitude of the experimental cross section for peak 5, particularly at small ϕ (ϕ < 5°). This is exactly what is found in Fig. 10. However when the PWIA-DFT results are scaled by a factor of 0.51, good agreement between the experimental measurements and theoretical MDs, for ϕ ≤ 5°, was found.17 This is also illustrated in Fig. 10 for the TZVP basis and BP-XC functional, but is equally valid for all the basis states and XC functionals studied. Hence the spectroscopic factor for the 1a1 MO at εf = 25.0 eV is S1a1 ≈ 0.51. Note that the agreement between the scaled PWIA-DFT results and the experimental MDs is still not perfect, the experimental cross section being larger in magnitude for ϕ > 5°. From our previous experience71 with tightly bound systems, i.e. inner valence and core states, this sort of behaviour is consistent with the PWIA providing an inadequate description for the ionisation mechanism. Under these circumstances14 a multi-centred distorted wave calculation (currently unavailable in molecules) would be necessary to correctly describe the collision dynamics. Consequently the lack of agreement for ϕ > 5° between 0.51 × PWIA-DFT 1a1 result and the experimental MDs was probably not a reflection of any serious limitation in the DFT basis states and XC functionals, rather it was a problem with the PWIA description of the reaction mechanism in this case.
Figures of the Wang et al.17 MDs for peaks 6 and 7 and the continuum are not plotted here, although in each case there is clear evidence for significant mixing of 1a1 and 1b2 spectral strength (see Table 3). Rather, in Fig. 11, we plot the MDs for the sum of the intensities under all of peaks 4–7 and the continuum (i.e. the cross section for the 1b2 + 1a1 orbitals), and the 1b2 + 1a1 MD PWIA-DFT theory results. For ϕ < 10° the level of agreement between theory and experiment is very good, while for ϕ > 10° the experimental cross sections are somewhat larger in magnitude than the corresponding calculations (see Fig. 11). Once again, we believe this discrepancy, at ϕ > 10°, is due to limitations with the PWIA. Nonetheless it is apparent from Fig. 11 that all the 1b2 and 1a1 spectral strength has been accounted for under peaks 4–7 and in the continuum.62 This is equivalent to saying that the spectroscopic factors for the respective 1b2 and 1a1 orbitals both add up to 1, namely the spectroscopic sum rule for these molecular orbitals of allene has been validated.8 This was, as discussed above, also found by Wang et al. for the 2e, 1e + 2b2 and 2a1 orbitals. All the HREMS binding energies and spectroscopic factors17 for each of allene's MO manifolds are summarised in Table 3.
| Orbital | HREMS | Manifold sum for S(f)j |
|---|---|---|
| 2e | 10.25 (∼1) | |
| 1e | 14.85 (∼1) | |
| 2b2 | 14.85 (∼1) | |
| 2a1 | 17.2 (∼1) | |
| 1b2 | 22.25 (∼0.67) | |
| 25.0 (<0.02) | ||
| 27.8 (∼0.10) | (∼1) | |
| 31.0 (∼0.09) | ||
| ≥33.5 (∼0.10) | ||
| 1a1 | 22.25 (∼0.09) | |
| 25.0 (∼0.51) | ||
| 27.8 (∼0.14) | (∼1) | |
| 31.0 (∼0.14) | ||
| ≥33.5 (∼0.10) |
Usually for EMS and HREMS studies in molecules, there were always experimental MDs, for one or two of the respective MOs, that could be used to make a clear distinction between the quality of the various basis sets and XC functionals that were employed in the DFT calculations.44 For allene, however, Wang et al. found that the situation was not so transparent. While it appeared, on the basis of the comparison between the experimental and theoretical MDs, that TZVP provided a better physical representation of the allene molecule than did either the DZVP or DZVP2 basis sets, it was less clear which of the non-local XC functionals (BLYP, BWP, BP or WP) was superior, although all were better than the LSD level. Indeed, while Wang et al.17 could have made a case that the BLYP/TZVP and BP/TZVP DFT basis sets and XC functionals led to MDs that were superior to those from the BWP/TZVP and WP/TZVP cases, it was impossible for them to distinguish between BLYP/TZVP and BP/TZVP simply on the MD comparisons alone. In this circumstance Wang et al. based their decision for the “optimum” DFT wave function of allene by relying on their past experience with other molecular systems15,16,40,70 to choose the appropriate XC functional. Under this criterion their “optimum” DFT wave function was BP/TZVP.
Wang et al.17 then took this “optimum” DFT-BP/TZVP basis set and used it to derive some of allene's molecular properties. These were then compared with independent experimentally determined values, in order to cross-check how well this optimum basis set was able to reproduce the molecular properties. In general, they found their calculations of molecular geometric properties using the BP/TZVP XC functional and basis set were in very good agreement with the independent measurements.57,58,63,66 Some of the results of Wang et al. are summarised in Tables 4 and 5. In particular, the carbon–carbon bond distance of 1.311 Å from their calculations agreed very well with the two available57,58 microwave spectroscopic method results of 1.308 and 1.309 Å, respectively. Their HCC bond angle of 120.962° was also in good agreement with that obtained by Maki and Toth,58 both being somewhat larger than the value from Hirota and Matsumara57 of 118.2°.
There have been a number of experimental determinations57,63,66 of the vibrational spectrum of allene. These results are summarised in Table 5, along with the results from the BP/TZVP calculation of Wang et al.. Their result for the ν4 mode lies comfortably within the range for the experimentally determined values,57,63,66 and is in particularly good accord with the value from Yang et al.66 Similarly, the results for the ν1,ν9 and ν11 modes of Hirota and Matsumara57 are also in quite good accord with the corresponding BP/TZVP results of Wang et al..
5.2 [1.1.1]Propellane (C5H6)
[1.1.1]Propellane (see Fig. 3b) is a quite remarkable hydrocarbon,39,72 with “inverted” geometries at the bridgehead carbon atoms, which consequently has been the subject of many experimental and theoretical studies of its properties.73 The structure,74 vibrational spectrum,75,76 total energy and strain energy,77 and heat of formation have all been investigated and a study into its low-energy electron impact spectroscopy has also been reported.78 The compound was found to be remarkably stable and to have a surprisingly short bridgehead C1–C3 bond length (160 pm), considering the extreme deviation from tetrahedral geometry and the anticipated steric strain. An excellent review of the chemistry of [1.1.1]propellane can be found in Levin et al.79The outervalence structure of [1.1.1]propellane has been previously studied with PES using He (I) radiation by Honegger et al.80 The Flinders group published the only previous EMS studies of [1.1.1]propellane15,81 which reported MDs for its complete valence electronic structure and experimental binding-energy spectra at two azimuthal angles, ϕ = 0° and 10°. Note that all the HREMS binding energies for each of [1.1.1]propellane's MO manifolds are summarised in Table 6.
| Orbital | EMS |
|---|---|
| 3a′1 | 9.7 |
| 1e″ | 11.3 |
| 3e′ | 12.6 |
| 1a′2 | 13.4 |
| 2e′ | 15.7 |
| 1a″2 | 17.0 |
| 2a′1 | 19.0 |
| 22.0 | |
| 1e′ | 26.0 |
| 29.0 | |
| 1a′ | 35.4 |
| 40.5 |
By comparing the experimental and theoretical MDs, for the relevant valence oribtals, Adcock et al.15 independently determined which of the SCF or DFT basis states they studied provided the most physically reasonable representation of the [1.1.1]propellane molecule. Adcock et al.15 utilised this optimum wave function to extract the chemically important molecular property information for [1.1.1]propellane and the state of hybridisation of the bridgehead carbon atoms.
Fig. 12 compares the experimental MD of Adcock et al.15 for the 3a′1 HOMO at εf = 9.7 eV, with a small selection of their PWIA-SCF and PWIA-DFT calculation results. It is clear from Fig. 12 that the measured MD for this state is strongly peaked at smaller values of p (or ϕ). Historically, with the development of EMS, those orbitals which are symmetric in coordinate space have high (e,2e) cross sections at small p.8 They are usually referred to as being “s-like” in nature. Thus the result of Fig. 12 implies that this HOMO is “s-like”. All of the theoretical PWIA-SCF and PWIA-DFT results are consistent with this observation, although it is apparent from Fig. 12 that the 6-31G Pople basis results in a MD which seriously underestimates the measured (e,2e) cross section at small p. On the other hand, both the BP/TZ94P and BSPP/TZ94P basis sets (not plotted) lead to a calculated (e,2e) cross section which is too large, compared to the experimental result, at small values of p (p ≈ 0.16 ℏ/a0). If the result of Adcock et al.15 for the BSPP/TZ94 basis calculation is scaled by their previously determined Green's function spectroscopic factor81 for the 3a′1 state (0.89), then the level of agreement between the experimental and theoretical MDs is good.
![The 1000 eV symmetric noncoplanar MD for the 3a′1 state of [1.1.1]propellane. The data of Adcock et al.15 (●) are compared against the results of their PWIA-SCF triple zeta basis (—), PWIA-SCF 6-31G Pople basis (- - -) and PWIA-DFT BSPP/TZ94 basis (— — —) calculations. Also shown is the result of the PWIA-DFT BSPP/TZ94 basis (-----) scaled by a factor of 0.89. Reproduced with permission of the American Chemical Society.](/image/article/2002/P2/b100700i/b100700i-f12.gif) | ||
| Fig. 12 The 1000 eV symmetric noncoplanar MD for the 3a′1 state of [1.1.1]propellane. The data of Adcock et al.15 (●) are compared against the results of their PWIA-SCF triple zeta basis (—), PWIA-SCF 6-31G Pople basis (- - -) and PWIA-DFT BSPP/TZ94 basis (— — —) calculations. Also shown is the result of the PWIA-DFT BSPP/TZ94 basis (-----) scaled by a factor of 0.89. Reproduced with permission of the American Chemical Society. | ||
Fig. 13 shows the experimental MD for the 2e′ orbital, at εf = 15.7 eV, of [1.1.1]propellane15 along with the corresponding theoretical PWIA-SCF and PWIA-DFT results. Clearly none of the calculations, for any of the basis sets considered, provided an (e,2e) cross section in agreement with the experimental result. This is particularly apparent for momenta less than 0.76 ℏ/a0, where the measured cross section starts to increase in value until reaching a maximum at p ≈ 0.16 ℏ/a0, while all the theoretical cross sections tend to 0 as p approaches 0.16 ℏ/a0. Note that in this case orbitals which are anti-symmetric in coordinate space have very small (0 if angular resolution effects are removed) (e,2e) cross sections as p → 0 ℏ/a0. Historically they are referred to as being “p-like” in nature.8 For p > 0.76 ℏ/a0 there is fair agreement between all the 2e′ orbital PWIA-SCF and PWIA-DFT calculation results and the experiment. Previously, Adcock et al.81 had found from their Green's function calculation that the 2a′1 orbital was severely split, with a significant 2a′1 spectroscopic strength at almost the same binding-energy as that of the 2e′ orbital. If we now allow for some 2a′1 flux with our 2e′ PWIA-DFT calculation (55% contribution allowed for here), using the BSPP/TZ94 basis, then very good qualitative agreement was found between theory and experiment for this combined MD (Fig. 13 for the 2e′ + 0.55 × 2a′1 orbitals MD). Obviously the level of agreement for the (e,2e) cross section, between the theory result that incorporates a 2a′1 contribution to the 2e′ orbital and the measured data is still not perfect, perhaps reflecting a limitation with the orbital of Adcock et al.15 Nonetheless this agreement is still quite good, with that analysis providing the first unequivocal supporting evidence for the Green's function calculation 2a′1 orbital spectroscopic strength splitting result of Adcock et al.81 Note that Adcock et al.15 provided similar analyses to that we have just given, for all the valence electronic states of [1.1.1]propellene. That paper should be consulted if more detail is required.
![The 1000 eV symmetric noncoplanar MD for the 2e′ state of [1.1.1]propellane. Legends are the same as in Fig. 12. Also shown is the result for the PWIA-DFT BSPP/TZ94 basis (—
—
—) when a (0.55) 2a′1 spectroscopic strength is added to that for the 2e′ orbital. Reproduced with permission of the American Chemical Society.](/image/article/2002/P2/b100700i/b100700i-f13.gif) | ||
| Fig. 13 The 1000 eV symmetric noncoplanar MD for the 2e′ state of [1.1.1]propellane. Legends are the same as in Fig. 12. Also shown is the result for the PWIA-DFT BSPP/TZ94 basis (— — —) when a (0.55) 2a′1 spectroscopic strength is added to that for the 2e′ orbital. Reproduced with permission of the American Chemical Society. | ||
The nature of the C1–C3 bridging bond in [1.1.1]propellane is rather controversial.82 It is not clear whether there is a bridging bond at all, and if so, how it can be described. Attempts to describe the character of the bridging bond of [1.1.1]propellane have concentrated on: orbital theories, either to describe the bonding with predictive models, or as a means of analysing ab initio wave functions; electron density83 or related quantities. The studies of hybridisation in [l.m.n]propellanes performed by several workers gave contradictory results for [1.1.1]propellane, the most-strained example.79,84–87
Newton and Schulman84 noted that “the electron density in the interbridgehead region is little different from that in bicyclo[1.1.1]pentane, a compound in which no formal bridgehead–bridgehead bond exists”. Computed electron-density difference maps also appeared to support this conclusion, since they showed a region of charge depletion between the bridgehead carbons, just as they do for the bicyclo calculations. A later orbital analysis of an ab initio wave function was performed by Jackson and Allen.88 They focused on the valence canonical MOs and decomposed them using an interaction scheme between a C2 fragment and the outer parts of the rings (CH2 groups). The HOMO was found to result from the in-phase combination of 2pσ orbitals on the bridgehead carbons, which has substantial density in the contributing regions. Jackson and Allen claimed that this orbital “contributed nil to holding C1 to C3” and instead ascribed the bridgehead bonding to a degenerate pair of orbitals that place the electron density off-axis.
In contrast, the analysis of the total electron density by Wiberg and co-workers77,83 concluded that there is a quantitative difference between the electron density in the bridging region of [1.1.1]propellane and of the analogous bicyclic species, bicyclo[1.1.1]pentane. The latter showed a minimum in the centre of the region while the former showed a bond point.82 In addition, in [1.1.1]propellane the value of the electron density (ρ) at the bond point83 is 0.203 a0−3, while in bicyclo [1.1.1]pentane it is only82 0.098 a0−3. Wiberg et al.77,83 found that the HOMO does contribute to the bonding of the bridgehead carbons. Indeed one-third of the total electron density is due to the HOMO, with its principal contribution, at the bond point being due to the overlap of 2s orbitals. The assignment by Jackson and Allen88 of the bonding as a result of off-axis density, and their claim that there is “very little charge density along the C1–C3 line of centres” is at variance with this observation of a bond point in the geometrical centre of the [1.1.1]propellane molecule.
The EMS experiments and DFT calculations of Adcock et al.15 on [1.1.1]propellane helped resolve this controversy. Their optimum basis was found to be BSPP/TZ94, which gave the best overall agreement between their experimental and theoretical MDs. Using this basis set they calculated that the interbridgehead carbon–carbon distance was 1.590 Å, in excellent agreement with the experimental value74 of 1.596 Å. In relation to the interbridgehead region of [1.1.1]propellane they estimated the electron density and subsequently the bond order (nb), midway between the two bridgehead carbons. Their bond order for the interbridgehead bond in [1.1.1]propellane of 0.70 was very similar to the Wiberg value83 of 0.73. They15 also calculated the bond order for the corresponding region in bicyclo [1.1.1]pentane as 0.40 (zero electron density gives a bond order of 0.21 in this analysis) indicating little or no interbridgehead bond in this compound. A summary of the important molecular property results from Adcock et al.,15 and a comparison of them with the results from other experiments and calculations, is given in Table 7.
| Experimental | Theoretical | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Propertya | IR75 | ED74 | BSPP/TZ9415 | 6-31G*82 | 6-31G*76 MP2 | 6-31G*75 MP2 | 6-311G87 MP2 | 6-31G*87 MP2 | 6-31G*75 MP3 |
| a Cb = bridgehead (axial) carbon atoms; Cn = methylene (equatorial) carbon atoms. b Mean value. c From electron density at the bond critical point. | |||||||||
| r(Cb–Cn)/Å | 1.522 ± 0.002 | 1.525 ± 0.002 | 1.518 | 1.502 | 1.514 | 1.515 | 1.521 | 1.525 | 1.514 |
| 1.522 | |||||||||
| 1.523 | |||||||||
| 1.531 | |||||||||
| 1.529 | |||||||||
| 1.526 | |||||||||
| 1.525 ± 0.004b | |||||||||
| r(Cb–Cb)/Å | 1.60 ± 0.02 | 1.596 ± 0.005 | 1.586 | 1.543 | — | 1.594 | 1.602 | 1.596 | 1.572 |
| r(C–H)/Å | — | 1.106 ± 0.005 | 1.094 | 1.075 | 1.088 | — | 1.087 | 1.106 | — |
| ∠Cb–Cn–Cb/deg | — | 63.1 ± 0.2 | 62.69 | — | 63.5 | — | — | — | — |
| 62.66 | |||||||||
| 62.67 | |||||||||
| 62.67 ± 0.01b | |||||||||
| ∠Cn–Cb–Cb/deg | — | 95.1 ± 0.1 | 95.81 | — | — | — | — | — | — |
| 95.78 | |||||||||
| 95.57 | |||||||||
| 94.99 | |||||||||
| 95.15 | |||||||||
| 95.46 ± 0.33b | |||||||||
| E/Eh | — | — | −193.967 | −192.691 | — | −193.350 | −193.511 | −193.375 | −193.370 |
| Cb–Cb bond order | |||||||||
| Mulliken | — | — | −0.11 | — | — | — | — | — | — |
| Mayer | — | — | 0.55 | — | — | — | — | — | — |
| nbc | — | — | 0.70 | 0.73 | — | — | — | — | — |
The nature of the hybridisation of the C1–C3 bond in [1.1.1]propellane was also examined by Adcock et al.15 Newton and Schulman84 found the hybridisation of the interbridgehead bond to be sp4.13. On the other hand, an NMR study by Jarret and Cusumano85 found the hybridisation at the bridgehead carbons to have a much higher p character (sp8.6–sp4.8) in the three bonds forming the three bridgehead methylene bonds and much larger s character (sp0.5) for the hybrids forming the interbridgehead bond. A more detailed description of these works can be found in the excellent review from Levin et al.79
The EMS studies of Adcock et al. contributed to the understanding of the character of bonding between the bridgehead carbon atoms and their hybidisation state. The 3a′1 HOMO has very strong “s-character”, due to strong s contributions from the bridgehead and methylene carbons. The bridgehead s contributions are in-phase (bonding). There is also a strong out-of-phase (bonding) bridgehead p contribution. The degenerate 1e″ orbitals have p character. There is essentially no s contribution to the bridgehead, and the p contribution is bonding. The 3e′ orbital has antibonding s and p contributions to the bridgehead, both of which are responsible for the p-like character of the 3e′ momentum profile. The 1a′2 orbital has essentially no contribution from the bridgehead carbons. The finding by Adcock et al.15 that the HOMO makes an important contribution to the interbridgehead bond is consistent with the result of Kar and Jug.89 The very “s-like” nature of the HOMO (Fig. 12) is not inconsistent with the observation of Jarret and Cusumano85 in respect to the hybrid nature of the interbridgehead bond.
5.3 Cubane (C8H8)
From the time of its successful synthesis in 1964 by Eaton and Cole,90,91 the structure and molecular properties of cubane (see Fig. 3c) have been studied extensively by both chemists and physicists.92,93 Excellent reviews on the state of our knowledge, with regard to cubane's structural and molecular properties, can be found by Eaton,92 Tsanaktsidis93 and Yildirim et al.94 Consequently only a brief précis of these results is presented here.Cubane belongs to the octahedral point group Oh and has cubic symmetry, although we note that in its crystalline form it is rhombohedral (a = 5.34 Å; α = 72.26°).95–97 Consistent with its highly symmetric structure is a notable vapour pressure92 (1.1 mm at 25 °C) and a significant density92 of 1.29 g cm−3. Its recommended93 C–C and C–H bond lengths of 1.571 and 1.109 Å, respectively, are taken from the work of Hedberg et al.98 The CCH and CCC bond angles are, however, less precisely known and, respectively, lie in the ranges99 123–127° and 89.3–90.5°. Additional properties, such as its strain energy93 (157.4 kcal mol−1) and 1H and 13C NMR spectra100,101 have also been reported. In the latter case it was found that cubane produced single resonances at 4.04100 and 47.3 ppm,101 respectively, while the sole 1H 13C–H coupling constant was determined by Della et al.101 as 153.8 Hz. Finally we note that experimental infrared (IR)102 and Raman spectroscopy103 studies into cubane's vibrational modes are available in the literature, as are extensive series of calculations on this same topic.104
Contrary to the situation just described for the physicochemical properties of cubane, experimental studies into the valence electronic structure of cubane have been more restricted. To the best of our knowledge they appear to be limited to the PES studies of Bodor et al.105 and Bischof et al.,106 and the HREMS study of Adcock et al.16 Of the two PES studies, by far the most detailed was that of Bischof et al.,106 who reported high-resolution He (Iα) and He (IIα) spectra. Eight of cubane's nine valence states were observed in this work, with assignments of the bands being made on the basis of ab initio STO-3G and MINDO/3 calculations and Koopmans' theorem. Note that we have not adopted the orbital nomenclature put forward by Bischof et al.106 Rather we have preferred that used in Schulman et al.,107 who numbered the symmetry labels within each irreducible representation with reference to the valence-shell orbitals only (i.e., those of carbon 2s and 2p and hydrogen 1s parentage). From a theoretical perspective, calculations into cubane's valence electronic structure are also quite limited. We note the MINDO/1,2 orbital energy results reported in Bodor et al.,105 the STO-3G and MINDO/3 orbital energy results given in Bischof et al.,106 the SCF-Xα, STO-3G, MINDO/3 and INDO orbital energy values reported by Schulman et al.107 and the SCF-DZ level orbital energies provided in Almlöf and Jonvik.108 A Green's function level calculation for cubane's orbital energies and spectroscopic factors, to the third order in the algebraic diagrammatic construction (ADC(3)) method,109 was also given in Adcock et al.16 Note that all the HREMS binding energies for each of cubane's MO manifolds are summarised in Table 8.
| Orbital | HREMS |
|---|---|
| 2t2g | 9.6 (∼1) |
| 1t2u | 9.6 (∼1) |
| 1eg | 13.7 (∼1) |
| 2t1u | 14.3 (∼1) |
| 1a2u | 15.6 (∼1) |
| 2a1g | 17.6 |
| 1t2g | 18.5 |
| 1t1u | 22.1 |
| 22.9 | |
| 1a1g | 27.4 |
| 28.3 | |
| 29.1 | |
| 30.3 | |
| 31.6 | |
| 32.1 |
Typical binding-energy spectra of cubane in the region 6–35 eV and at E = 1000 eV are given in Fig. 14. In the same manner as described earlier, MDs for all of the respective valence orbitals (2t2g, 1t2u, 1eg, 2t1u, 1a2u, 2a1g, 1t2g, 1t1u, 1a1g) of cubane were derived by Adcock et al.16
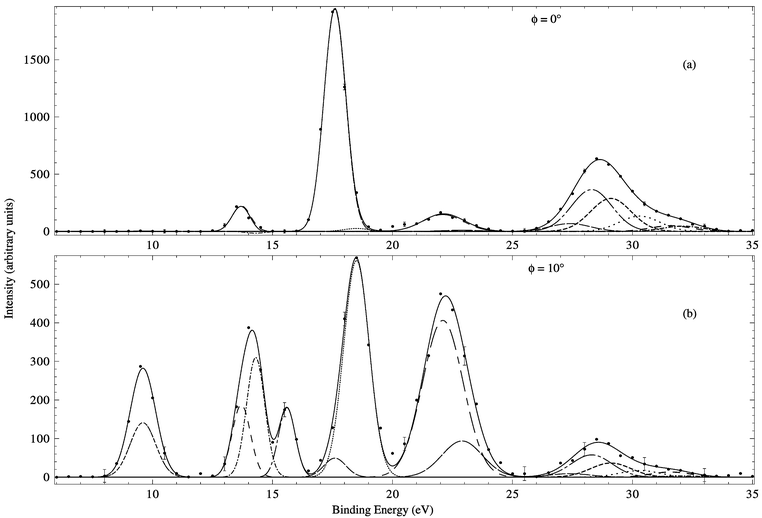 | ||
| Fig. 14 Typical binding-energy spectra from our 1000 eV noncoplanar symmetric EMS investigation into cubane. The curves show the fits to the spectra at (a) ϕ = 0° (p ≈ 0.09 ℏ/a0) and (b) ϕ = 10° (p ≈ 0.75 ℏ/a0) using the known energy resolution. Reproduced with permission of the American Chemical Society. | ||
In Fig. 15a we compare the experimental MD of Adcock et al.16 for the degenerate HOMOs, 2t2g + 1t2u at εf = 9.6 eV, of cubane with the results from their PWIA-LSD calculations for each of the seven DFT basis sets they considered. It is apparent from Fig. 15a that the TZ94P basis functions provide totally inadequate representations of the 2t2g + 1t2u orbitals, particularly for momenta ϕ < 12°. Specifically, TZ94P leads to an initial peak in the MD, at around ϕ = 8°, that is not seen experimentally or in the other PWIA-DFT/LSD calculations. A somewhat unexpected feature, compared to our usual8 experience, is that these “p-like” 2t2g and 1t2u outer valence orbitals have a peak in the MD at a remarkably high value of ϕ and have virtually no intensity (see also Fig. 14) at ϕ = 0°. This very interesting extreme “p-like” behaviour of the 2t2g + 1t2u orbitals may also point to interesting hybridisation. Traditionally, strained cyclic molecules are seen as having “normal” sp3-hybridised carbons, and the unusual carbon skeleton bond angles are explained by assuming the sp3 hybrid orbitals are “bent”. The results of Adcock et al.16 suggest that the HOMOs are almost purely “p-like” in character and so are better explained by an spn hybrid where n is large. This makes intuitive sense as the three p orbitals on carbon are orthogonal, just like the skeletal carbon bonds. Thus perhaps cubane has C–C bonds which are formed essentially by σ overlap of “pure” p orbitals. The s orbitals would then go mainly to forming C–H bonds. Theoretical studies of cubane by Schulman and co-workers107 are consistent with this chemically intuitive approach and provide a rationale for the extremely “p-like” MD observed for the HOMOs. They found that, because of its high symmetry, cubane represents an unusual case of a molecule which has a molecular orbital which is either solely C–C bonding or solely C–H bonding. In a minimal basis set treatment the outer valence 1eg and 1t2u (HOMO) orbitals are symmetry determined combinations of 2p orbitals with no 2s admixture. The 2t2g (HOMO) orbital is a mixture of C–C and C–H bonds with C–C dominating. Finally, we note that the results in Fig. 15a suggest that the experimental spectroscopic factors for both the respective 2t2g and 1t2u orbitals are ≈1. This observation is entirely consistent with the ADC(3) spectroscopic factor calculations for these orbitals from Adcock et al.16
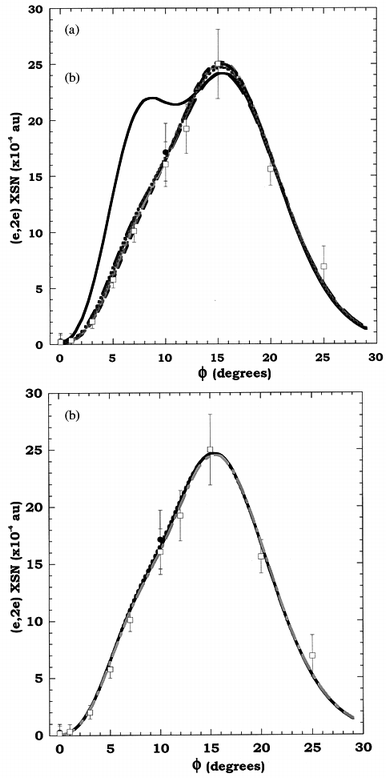 | ||
| Fig. 15 (a) The 1000 eV symmetric noncoplanar MD for the 2t2g + 1t2u HOMOs of cubane. The data of Adcock et al.16 for runs A (●) and B (□) are compared against the results of their PWIA-DFT/LSD calculations: (— -) DZ94, (— — —) DZ94P, (- - -) DZVP, (—— - -) DZVP2, (— - - - —) TZ94, (——) TZ94P and (- - - - - -) TZVP. Reproduced with permission of the American Chemical Society. (b) The 1000 eV symmetric noncoplanar MD for the 2t2g + 1t2u HOMOs of cubane. The data of Adcock et al.16 for runs A (●) and B (□) are compared against the results of their PWIA-DFT/TZVP calculations at the LSD level (- - -), BLYP level (——) and BP level (— - -). Reproduced with permission of the American Chemical Society. | ||
Fig. 15b shows that the PWIA-DFT MD result with TZVP basis functions is largely insensitive to whether LSD, BP or BLYP non-local functional corrections are used. This observation also holds for each of the other six DFT basis functions Adcock et al.16 employed in their study, although for the sake of clarity they are not illustrated in Fig. 15b. In addition, this insensitivity of the calculated MDs to whether LSD or GGA (BP or BLYP) levels were employed was also found to occur for the remaining outer valence (1eg, 2t1u and 1a2u) orbitals. Finally, we note from Fig. 15b the excellent agreement between the PWIA-DFT LSD, BP, BLYP/TZVP MD results and the experimental MD result over all measured ϕ.
In Fig. 16 we compare the experimental MD for the first of the inner valence orbitals, the 2a1g orbital at εf = 17.6 eV, with the results from Adcock et al.'s PWIA-DFT LSD calculations for each of the seven DFT basis sets they studied. Unlike those for the outermost valence orbitals of cubane, the 2a1g MD is clearly “s-like” in nature, having a strong maximum in the cross section at ϕ = 0°. We note, however, the broad secondary peak in this cross section with a much weaker maximum near ϕ = 13.5°, indicating some “p-like” contribution to the MD for the 2a1g orbital. It is apparent from Fig. 16 that the PWIA MDs for both the LSD/DZVP and LSD/DZVP2 basis states somewhat underestimate the magnitude of the experimental MD for ϕ < 5°. In addition, the failure of the LSD/TZ94P basis to provide an adequate representation for the orbital in question is manifest, particularly for ϕ < 10° where the calculated cross section is often a factor of 2.5 times larger than that observed experimentally.
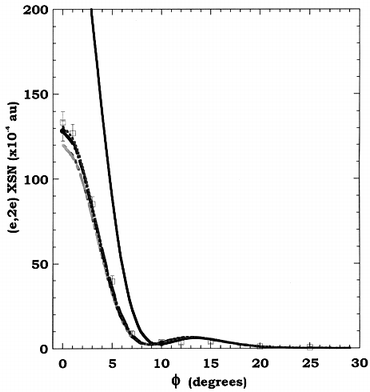 | ||
| Fig. 16 The 1000 eV symmetric noncoplanar MD for the 2a1g orbital of cubane. The legend is the same as that for Fig. 15a. Reproduced with permission of the American Chemical Society. | ||
The descriptions we provided above, in our detailed comparison of the experimental and theoretical MDs of Adcock et al.,16 for a small but representative subset of the valence orbitals of cubane, holds equally well for the remaining 1eg, 2t1u, 1a2u, 1t2g, 1t1u and 1a1g orbitals. Consequently we do not specifically discuss the MD results for these latter valence orbitals except to highlight two further points. First, the experimental spectroscopic factors for the respective 1eg, 2t1u and 1a2u outer valence orbitals were all found by Adcock et al. to be ∼1, in good accord with their corresponding ADC(3) calculation results.16 Second, for the 1t2g and 1t1u orbitals we notice a suggestion that the PWIA calculation with the BP/TZVP basis and non-local correction provided a somewhat better description of the respective experimental MDs than did either of the corresponding LSD/TZVP or BLYP/TZVP results.
If we now briefly summarise the results of Adcock et al.,16 as embodied in Figs. 15 and 16, we note that there is little to separate the quality of the description of the experimental MDs, for all the valence orbitals, provided by both the PWIA-DFT LSD/TZVP and LSD/TZ94 calculations. After very careful consideration of their results, Adcock et al.16 concluded that of these two basis sets the LSD/TZVP calculation provided a marginally superior description of the experimental results. In addition, the total energy of their BLYP/TZVP wave function16 was −309.4009 Eh, while that for their BLYP/TZ94 wave function was −309.3138 Eh. As the total energy of the TZVP result is lower (more negative) than that of the TZ94 result, we can surmise that it provides the most stable configuration for the cubane molecule for the basis states they considered. Consequently when Adcock et al. coupled this latter result with that afforded by their comprehensive comparison between the experimental and theoretical MDs, they concluded that their16 “optimum” wave function for the cubane molecule was TZVP.
In general, Adcock et al.'s16 calculations of molecular geometric properties using the TZVP basis set were in excellent agreement with experimentally determined molecular properties, and compare favourably with the results from other MO calculations. Their results are summarised in Table 9. In particular, the carbon–carbon bond distance of 1.575 Å from their calculations16 agreed very well with the two most accurate experimentally determined values of 1.571 Å from electron diffraction110 and microwave spectroscopic methods.111 This compares with 1.551 Å from a lower accuracy X-ray diffraction study.95 The carbon–hydrogen bond lengths were 1.098 Å from the TZVP calculations, compared with 1.097 Å by microwave spectroscopy,111 1.098 Å by electron diffraction,110 1.040 Å from the lower accuracy X-ray diffraction study, and the recommended value of 1.109 Å from Tsanaktsidis.93 Adcock et al.'s calculations yielded CCC bond angles of 90.00° and CCH bond angles of 125.25° also in excellent agreement with the electron diffraction and microwave values. Miaskiewicz and Smith104 published a theoretical study of cubane using the 6-31G* basis set and several DFT functionals. Their104 geometries derived from BPW91 functionals were similar to Adcock et al.'s16 in terms of agreement with accurate microwave and electron diffraction geometries, with other functionals yielding inferior results.
Della et al.101 measured the NMR spectra of cubane and derived the 13C–H one-bond coupling constant. Their experimentally derived value of 153.8 Hz is in very good agreement with the Adcock et al.16 theoretical values of 153.6–154.7 Hz, using the TZVP basis set and the IGLO method, and 154.1–155.0 Hz using the LORG method. Eaton and Cole91 also measured the NMR spectra of cubane and obtained a value of 155 Hz for this coupling constant.
There have been a number of experimental determinations of the vibrational spectrum of cubane, both infrared and Raman. Della and his colleagues first measured the solution (CS2 and CCl4) and solid-phase infrared and Raman spectra.103 More recently Pine et al.112 have used a tuneable laser to investigate the vibrational spectrum of cubane and derived a quadratic force field for the molecule. There have also been several theoretical studies of the vibrational spectra of cubane and its isotopically substituted forms, using various levels of MO theory. Jursic113 employed hybrid and gradient-corrected DFT methods to compute the infrared and Raman spectra of cubane. He found that the B3LYP method gave the best agreement with experiment, especially after correcting the theoretical spectrum using empirical correlation factors to improve the agreement with experiment. Miaskiewicz and Smith104 compared the performance of various DFT functionals with, and without, additional frequency scaling for calculating the cubane vibrational frequencies. They found that BPW91, BLYP and B3LYP functionals gave good agreement with experimental spectra and also acceptable geometries for cubane, with the BPW91 functional being the best compromise. They note that the SVWN functional, which is the “cheapest” of all DFT methods, gave a very good spectral prediction, albeit with worse prediction of geometry.
Table 10 shows the vibrational frequencies calculated using the TZVP basis from Adcock et al.,16 compared with the experimentally measured infrared and Raman spectra of cubane. The calculated intensities of the transitions from their TZVP basis are also in reasonable agreement with the observed experimental infrared spectrum of cubane,103,112 as Table 11 illustrates. The TZVP calculations of Adcock et al.16 gave frequencies of the vibrational modes of cubane in quite good agreement with experiment. After applying small empirical scaling factors (of C–C–C bend, 1.053; C–C stretch, 1.070; C–C–H bend, 1.040; C–H stretch, 0.974) to the frequencies, their predictions were considerably improved. These scaling factors were similar to those derived by Miaskiewicz and Smith104 and Jursic.113 We note that the Adcock et al.16 calculations gave results of comparable accuracy to those of Miaskiewicz and Smith.104 The TZVP basis set is thus a good compromise where an accurate prediction of geometry and vibrational spectra is required.
| Expt | Adcock et al.16 | ||
|---|---|---|---|
| Assignment102 | Infrared102/Raman103 | TZVP | Scaled TZVP |
| a Assignment of cubane vibrational modes follows the work of Vlahacos et al.102 The C–C–H bending mode can be alternatively described as C–H wagging. | |||
| C–C–C bend | 617 | 523 | 582 |
| C–C–C bend | 667 | 623 | 656 |
| C–C stretch | 821 | 736 | 788 |
| C–C stretch | 829 | 783 | 838 |
| C–C stretch | 852 | 794 | 850 |
| C–C stretch | 899 | 821 | 879 |
| C–C stretch | 1001 | 970 | 1038 |
| C–C–C bend | 1030 | 1003 | 1056 |
| C–C–H bend | 1036 | 1022 | 1063 |
| C–C–H bend | 1078 | 1037 | 1078 |
| C–C–H bend | 1083 | 1040 | 1082 |
| C–C–H bend | 1130 | 1096 | 1140 |
| C–C–H bend | 1183 | 1121 | 1166 |
| C–C–H bend | 1235 | 1163 | 1210 |
| C–H stretch | 2965 | 3046 | 2966 |
| C–H stretch | 2972 | 3054 | 2974 |
| C–H stretch | 2990 | 3060 | 2981 |
| C–H stretch | 2994 | 3077 | 2997 |
The molecular electrostatic potentials from their TZVP calculations16 are symmetric. In addition these calculations also indicated negative charge on the carbon atoms (Mulliken net atomic charge −0.116, Löwdin net atomic charge −0.101, charges fitted to electrostatic potential −0.032) and balancing positive charges on the hydrogen atoms. Adcock et al.16 also investigated the electron density in the carbon–carbon region of cubane and found that the maximum electron density lies off the carbon–carbon axis by approximately 0.05 Å, consistent with the “bent bond” hypothesis for highly strained molecules. The off-axis electron density implies a bond angle of approximately 93.5° i.e., the electron density in the bond forms an angle of approximately 3.5° with the carbon–carbon axis.
Adcock et al.16 carried out a study analogous to that of Wiberg and colleagues,77 and their earlier15 [1.1.1]propellane work, to estimate the electron density at the bond critical point. They obtained a value of ρb = 0.224 a0−3. Adcock et al. then employed Wiberg's empirical method to calculate bond orders from electron densities at the bond critical points derived from the TZVP basis functions. The electron densities at the bond critical points of the model compounds ethane, ethene, ethyne and benzene (bond orders of 1.0, 2.0, 3.0 and 1.5 respectively) were used to determine the constants in the relation between bond order n and the bond critical point densities ρb (eqn. (9)).
| n = exp{7.075(ρb − 0.2377)} | (9) |
This relationship yielded a bond order for the carbon–carbon bonds in cubane of 0.91, a value in good accord with that found by Wiberg et al.77 (n = 0.95) with their HF/6-31G* basis. Adcock et al.16 also calculated the bond order of the carbon–carbon bonds using Mulliken and Mayer population analysis. The Mayer bond order of 0.95 was in good agreement with their value. The Mulliken value of 0.58, on the other hand, again reflects the well-known deficiencies of this method of orbital decomposition.
5.4 Norbornadiene (C7H8)
The electronic structure of norbornadiene (bicyclo[2.2.1]heptadiene) (see Fig. 3d) has been of considerable interest as it is the prototypical molecule for the study of through-space and through-bond interaction, concepts originally introduced by Hoffmann and colleagues59,60,114 and incorporated into an SCF scheme by Heilbronner and Schmelzer.61 The assignment of the first two ionic states, which arise from the ejection of an electron from the symmetric (π+) and antisymmetric (π−) linear combination of the two ethylenic π bond orbitals: π± = πa ± πb, would give information on whether the through-space or through-bond interaction dominates. This follows because if the through-space interaction were to dominate, π− would be expected59,60,114 to be the HOMO, while if the through-bond interaction were to dominate, π+ would be expected59,60,114 to be the HOMO.The unique property of EMS of being able to unambiguously identify the symmetry of an orbital, from its measured MDs for binding-energy selected electrons, thus makes it an ideal technique to definitively determine the dominant interaction in the outermost π orbitals for norbornadiene (NBD). Specifically, if the π− antisymmetric orbital was the HOMO we would expect to observe an experimental MD for the HOMO that had small intensity at low momentum,8 whereas if the π+ symmetric orbital was the HOMO the converse would be true, i.e. its experimental MD would have appreciable intensity at low momentum.8
Previous PES studies into the valence electronic structure of NBD include the He (I) measurements from Bischof et al.115 and the He (II) measurements from Bieri et al.116 These results were interpreted by von Niessen and Diercksen,117 using an ab initio many-body Green's function method, to indicate that the HOMO is the 5b2 (π) orbital (which is essentially the π− combination). Consequently, von Niessen and Diercksen117 argued that it is the through-space interaction which dominates between the orbitals πa and πb in NBD. This result was consistent with the earlier conclusion of Heilbronner and Martin118 although, as noted by both von Niessen and Diercksen117 and Galasso,119 the ordering of the orbital energies is not uniformly reproduced by the calculations. Hence we believe that at that time the definitive confirmation was still outstanding.
In light of this situation Takahashi et al.120 undertook the pioneering, low-resolution, EMS study on NBD. This work was conducted at a total energy of 1200 eV and an energy resolution of 1.5 eV (FWHM). Unfortunately, the HOMO and the next-highest-occupied-molecular-orbital (NHOMO) in NBD are only separated by 0.85 eV,115,116 and so they could not resolve them in their binding-energy spectra.120 In addition, with this broad energy resolution, contributions from the next highest 2a2 orbital to the HOMO and NHOMO flux could also not be ruled out. To try and circumvent these difficulties Takahashi et al.120 used a spectral deconvolution procedure, but the uniqueness of this procedure is debatable in this case as is reflected by the scatter in their data120 for the HOMO and NHOMO MDs. Thus while their120 results were consistent with the conclusion of von Niessen and Diercksen,120 for the dominance of the through-space interaction, they were by no means definitive. Consequently Mackenzie-Ross et al.121,122 made new HREMS measurements and performed PWIA-DFT calculations, in an attempt to establish beyond doubt whether the through-space or through-bond interaction is dominant in NBD.
Typical binding-energy spectra for the complete valence shell of NBD in the region εf = 6–33 eV and at a total energy of 1500 eV are given in Figs. 17a (ϕ = 0°) and 17b (ϕ = 10°).121 Also shown in these spectra are enhancements for the binding-energy regions pertaining to the HOMO and NHOMO. Figs. 17a and b clearly show that peaks 1 and 2 (HOMO and NHOMO respectively) are resolvable from one another and that both these peaks are well separated from the 2a2 orbital117 (peak 3).
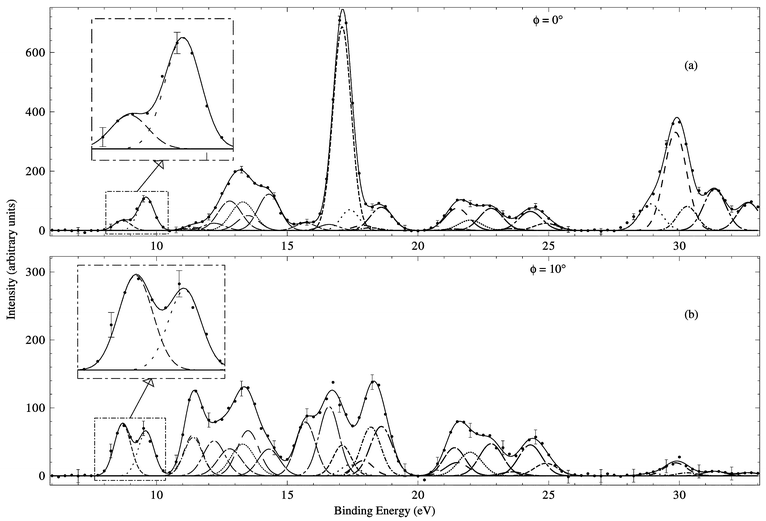 | ||
| Fig. 17 Typical binding-energy spectra from the 1500 eV noncoplanar symmetric HREMS into norbornadiene of Mackenzie-Ross et al.121 The curves show the fits to the spectra at (a) ϕ = 0° and (b) ϕ = 10° using the known energy resolution. Reproduced with permission from Elsevier Science. | ||
The binding-energy spectra were then analysed with the least squares-fit deconvolution technique discussed previously.22 This deconvolution analysis allowed Mackenzie-Ross et al.122 to derive the required MDs for all the respective valence orbitals of NBD. From a theory perspective, Mackenzie-Ross et al.121,122 used three basis sets in their DFT computations. These basis sets were DZVP, DZVP2 and TZVP. Note that the DFT calculations they performed121,122 used the GGA methods in the geometry optimisations. Theoretical MDs were then calculated in the usual manner within the PWIA framework.
The MDs for the first two peaks (HOMO and NHOMO) in their121 binding-energy spectra (see Figs. 17a and b) are given, respectively, in Figs. 18 and 19. It is clear from Figs. 18 and 19 that these MDs differ significantly from one another. Note that the results from Mackenzie-Ross et al.121 were for two independent runs (A and B) and that both are plotted in these figures. Further note that in all cases for both orbitals they are consistent with each other. Also plotted in these figures are the corresponding (1200 eV total energy) MDs from Takahashi et al.120 and the MD results from Mackenzie-Ross et al.'s PWIA-DFT computations for the 5b2 and 7a1 orbitals. The 5b2 orbital PWIA MD results, with BP/DZVP, BP/DZVP2, BP/TZVP and BLYP/TZVP basis sets and XC functionals, reproduce very well their121 experimental MD for the HOMO (see Fig. 18).This is a strong indication that the HOMO is the π antisymmetric orbital. Similarly, the 7a1 orbital PWIA MD results, again with BP/DZVP, BP/DZVP2, BP/TZVP and BLYP/TZVP DFT basis sets and XC functionals, also reproduce very well their121 experimental MD for the NHOMO (see Fig. 19). The only exception to this observation is that their 7a1 theory MD with BP/DZVP basis tends to somewhat underestimate the magnitude of the (e,2e) cross section for p ≤ 0.4 ℏ/a0, thereby suggesting a limitation with the accuracy of BP/DZVP. Nonetheless, the generally good agreement between their experimental NHOMO MD and their theoretical 7a1 MDs is important evidence for the NHOMO being the π+ symmetric orbital.
 | ||
| Fig. 18 The 1500 eV symmetric noncoplanar MD for the 5b2 HOMO of norbornadiene. The data of Mackenzie-Ross et al.121 for Run A (●) and Run B (△) and the earlier data of Takahashi et al.120 (□) are compared against the results of their121,122 PWIA-DFT calculations: (— —) BLYP/TZVP, (——) BP/TZVP, (- - - - - -) BP/DZVP2 and (· —) BP/DZVP. Acronyms are defined in the text. Reproduced with permission from Elsevier Science. | ||
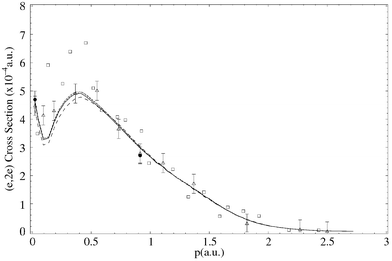 | ||
| Fig. 19 The 1500 eV symmetric noncoplanar MD for the 7a1 NHOMO of norbornadiene. The legend is the same as that for Fig. 18. Reproduced with permission from Elsevier Science. | ||
While the earlier MD data of Takahashi et al.120 exhibited more scatter than that of Mackenzie-Ross et al.,121 and have larger uncertainties (not plotted) than the Mackenzie-Ross et al. results, the trend is clear. Namely, for both the HOMO and NHOMO the momentum distribution results of Takahashi et al.120 are in good quantitative accord with those from Mackenzie-Ross et al.121 Another interesting observation from the HOMO and NHOMO MDs is that for both these orbitals there is a local minimum in the PWIA-DFT results for p ≈ 0.1 ℏ/a0. The experimental data of Mackenzie-Ross et al.121 provides some support for the existence of these local minima, which we believe arise due to electron correlation effects.
The most exciting result from Figs. 18 and 19 is, however, the following. It is clear from these figures that the (e,2e) cross section for the HOMO is relatively small at p ≅ 0 ℏ/a0 (see Fig. 18), while that of the NHOMO (Fig. 19) has significant strength in the lower momentum region. Since only totally symmetric orbitals can have an appreciable (e,2e) cross section8 at p ≅ 0 ℏ/a0, our result in Fig. 19 unequivocally demonstrates the NHOMO is the symmetric π+ orbital. Consequently, consistent with the HOMO having a relatively small (e,2e) cross section at p ≅ 0 ℏ/a0, the HOMO must be the antisymmetric π− orbital. Hence the measurements of Mackenzie-Ross et al.121 definitively confirmed the dominance of the through-space bond interaction between πa and πb in NBD.
The remaining 16 valence electronic state MDs of NBD were reported by Mackenzie-Ross et al.,122 with an example for the 3b2 orbital being given in Fig. 20. In this case it is apparent from Fig. 20 that the experimental MD is best reproduced by the PWIA-DFT calculation with either BLYP/TZVP or BP/TZVP XC functionals and basis. Indeed, when they made a comprehensive comparison between all their experimental and theory MDs, Mackenzie-Ross et al.122 found that the best overall description was provided by the BP/TZVP basis.
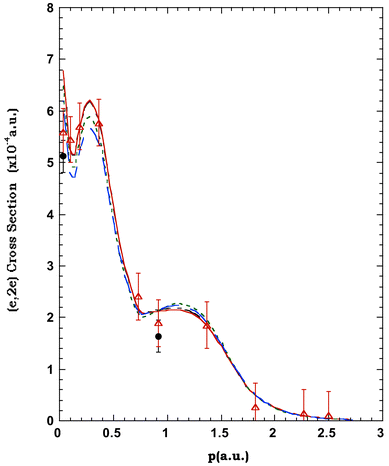 | ||
| Fig. 20 The 1500 eV symmetric noncoplanar MD for the 3b2 orbital of norbornadiene. The legend is the same as that for Fig. 18. | ||
In general Mackenzie-Ross et al.122 found that their calculations of molecular geometries were in very good agreement with other experimentally determined geometries123,124 and also they compared favourably with the results from other MO calculations.125–127 Some of these results are summarised in Table 12. In particular, the bridgehead carbon–carbon distance of 1.563 Å from their122 calculations was in excellent agreement with the two most accurate experimental values of 1.557 Å from a Fourier transform microwave (FTMW) study123 and a value of 1.571 Å from an electron diffraction (ED) study.124 The carbon–carbon double bond lengths were 1.341 Å from their BP/TZVP calculations,122 compared with 1.3362(30) Å from FTMW and 1.3387(12) Å from ED. The non-bridgehead single bonds were slightly overestimated by BP/TZVP compared with experiment (see Table 12), but this was a smaller error than that from earlier SCF 3-21G calculations,125,126 and an SCF 6-31G* calculation.127 The distance between the two double bonds was particularly well reproduced by Mackenzie-Ross et al.,122 with the C2⋯C6 distance from their BP/TZVP of 2.487 Å compared with the experimental distance of 2.473 Å (FTMW) and 2.462 Å (ED). Trends in the C–H bond lengths determined in the FTMW study123 were reproduced by the BP/TZVP calculation of Mackenzie-Ross et al.,122 with experimental values typically being 0.007 Å smaller than their theory results. It is also apparent from Table 12 that the respective bond angles of NBD were well reproduced by their122 calculation. Finally, even though it is more of an electronic rather than a geometric property, we note that the small dipole moment of NBD is reproduced quite well by the BP/TZVP calculation of Mackenzie-Ross et al.122 They obtained a value of 0.082 D, compared with the very accurate FTMW value123 of 0.05866(9) D.
| Parameter122 | FTMW123 | ED124 | BP/TZVP122 | 3-21G125,126 | 6-31G*127 |
|---|---|---|---|---|---|
| r(C1–C2)/Å | 1.5304(31) | 1.5332(14) | 1.546 | 1.550 | 1.5395 |
| r(C1–C7)/Å | 1.5567(28) | 1.5711(31) | 1.563 | 1.566 | 1.5505 |
| r(C2–C3)/Å | 1.3362(30) | 1.3387(12) | 1.341 | 1.319 | 1.3192 |
| r(C1–H)/Å | 1.0903(13) | 1.1094(47) | 1.097 | 1.076 | — |
| r(C2–H)/Å | 1.0809(13) | 1.0896 | 1.090 | 1.069 | — |
| r(C7–H)/Å | 1.0954(12) | 1.1094(47) | 1.101 | 1.081 | — |
| ∠C1C2C3/° | 107.13(9) | — | 107.14 | 107.5 | — |
| ∠C1C7C4/° | 91.90(17) | 92.2(4) | 92.256 | 92.0 | 91.87 |
| ∠C2C1C6/° | 107.58(25) | — | 107.06 | 106.2 | 107.45 |
| ∠C2C1C7/° | 98.30(14) | — | 98.37 | 98.3 | 98.31 |
| ∠C7C1H/° | 117.66(26) | — | 117.82 | 118.2 | — |
| ∠C3C2H/° | 127.84(10) | 125.2(14) | 127.78 | 128.1 | — |
| ∠HC7H′/° | 111.99(14) | 114.7(30) | 110.9 | 111.7 | — |
| d(C2⋯C6)/Å | 2.473 | 2.462 | 2.487 | — | — |
| μ/D | 0.05866(9) | — | 0.082 | — | — |
Similar to [1.1.1]propellane and cubane, Mackenzie-Ross and colleagues also investigated, using BP/TZVP, to determine the electron density in the carbon–carbon region of NBD. Here they obtained values at the bond critical point of ρb = 0.3282 a03 for the double bonds and 0.2266 a03 for the single bonds. Again employing the method of Wiberg and colleagues,77 this electron density translated into a carbon–carbon bond order in NBD of 1.90 for the double bonds and 0.924 for the single bonds. Mackenzie-Ross et al.122 also calculated the corresponding bond order of the carbon–carbon bonds using Mulliken and Mayer population analysis. The Mayer bond order of 1.90 for the double bonds and 0.95 for the single bonds was in good agreement with their values. The Mulliken value of 1.32 for the double bonds and 0.73 for the single bonds was deficient, again reflecting the limitations of this method of orbital decomposition.
6 Conclusions and future prospects
We have reviewed high-resolution electron momentum spectroscopy with particular emphasis on its application to allene, [1.1.1]propellane, cubane and norbornadiene. The utility of the technique, when combined with density function theory or SCF calculations, to elucidate the valence electronic structure and chemical bonding, as well as provide a wide-range of accurate data for important molecular properties, of organic molecules was established. In the process of this, the ability of electron momentum spectroscopy in general, and high-resolution electron momentum spectroscopy in particular, to a priori test the quality of the respective theoretical basis states under study was highlighted, an “optimum” density functional basis and exchange-correlation functional to represent each molecule being determined. As a general observation for the molecules we have thus far studied, it would appear that the BP/TZVP exchange correlation functional and basis provides the best representation for the respective measured MDs. In addition, molecular property information derived from it (BP/TZVP) also appears to be in generally quite good agreement with corresponding values from independent measurements.The future of high-resolution EMS looks bright. At Flinders we are preparing to collaborate with Gleiter's group at Heidelberg to study some of the stelladione hydrocarbons e.g. 2,6-stelladione (tricyclo[3.3.0.03,7]octane-2,6-dione).128,129 HREMS measurements, coupled with DFT calculations, when combined with precision PES measurements from the Heidelberg group, should provide detailed information on the repective valence electronic structures of these species. Indeed, the general application of the technique to larger and biologically interesting molecules5 is a trend that looks set to accelerate. From a technical perspective, rather than employing electron-monochromators, better energy-resolution can also be obtained with laser-based photo-electron sources. Dorn et al.130,131 have already used a negative electron affinity GaAs based spin-polarised electron source for (e,2e) studies with atoms. They found a significant improvement in their energy resolution to of the order of 0.5 eV (FWHM). For energy distributions of equivalent widths, the photoemission sources give currents which can be some ten or more times greater than those obtained using thermionic emitters coupled with monochromators.3 In addition, the possibility of detecting the molecular ions in triple coincidence with the scattered and ejected electrons, opens the way for oriented-molecule EMS studies. This exciting development would need to be accompanied by both the installation of multivariable detectors to gather data simultaneously over a range of energies and momenta, and the use of a supersonic nozzle and differential pumping techniques in order to increase the target molecular density. With these developments the triple coincidence experiment becomes feasible, in terms of workable count rates, and spherical averaging (see eqn. (7)) is removed. The consequence of this is that the measured cross section would then represent a significantly more stringent test for both the quality of the basis states employed in the calculation and the reaction mechanism description. This in turn could promote the development of more accurate basis states and XC functionals and ultimately, to the derivation of more exact molecular property information for the molecule under study.
7 Appendix
Definitions of acronyms are given in Table 13.| ADC(3) | Third order algebraic diagrammatic construction |
| AOs | Atomic orbitals |
| BP | Becke–Perdew exchange correlation functional |
| BWP | Becke–Wang–Perdew exchange correlation functional |
| BLYP | Becke–Lee–Yang–Parr exchange correlation functional |
| CI | Configuration interaction |
| DFT | Density functional theory |
| DZ | Double zeta |
| DZVP | Double zeta valence polarisation |
| EMS | Electron momentum spectroscopy |
| FWHM | Full-width-at-half-maximum |
| GGA | Generalised gradient approximation |
| HF | Hartree–Fock |
| HOMO | Highest-occupied-molecular-orbital |
| HREMS | High-resolution electron momentum spectroscopy |
| LDA | Local density approximation |
| LSD | Local spin density |
| MDs | Momentum distributions |
| MOs | Molecular orbitals |
| NBD | Norbornadiene |
| NHOMO | Next-highest-occupied-molecular-orbital |
| NMR | Nuclear magnetic resonance |
| PES | Photoelectron spectroscopy |
| PWIA | Plane wave impulse approximation |
| SCF | Self-consistent-field |
| TZ | Triple zeta |
| TZVP | Triple zeta valence polarisation |
| VWN | Vosko–Wilk–Nusair local spin density level correlation energy functional |
| WP | Wang–Perdew exchange correlation functional |
| XC | Exchange-correlation functional |
8 Acknowledgements
There are many people at Flinders, both past and present, who have contributed to the development of high-resolution electron momentum spectroscopy and we acknowledge their contribution. We thank Ms Marilyn Mitchell for typing this manuscript and Dr Laurence Campbell for his assistance in preparing some of the figures.9 References
- H.-D. Martin and B. Mayer, Angew. Chem., Int. Ed. Engl., 1983, 22, 283 CrossRef.
- M. N. Paddon-Row and K. D. Jordan, in Modern Models of Bonding and Delocalisation, VCH Publishers, New York, 1988, ch. 3 Search PubMed.
- E. Weigold, Aust. J. Phys., 1998, 51, 751 Search PubMed.
- I. E. McCarthy and E. Weigold, Rep. Prog. Phys., 1991, 54, 789 CrossRef CAS.
- Y. Zheng, J. J. Neville and C. E. Brion, Science, 1995, 270, 786 CAS.
- M. A. Coplan, J. H. Moore and J. P. Doering, Rev. Mod. Phys., 1994, 66, 985 CrossRef CAS.
- V. G. Neudatchin, Yu. V. Popov and Yu. F. Smirnov, Phys.-Usp., 1999, 42, 1017 CrossRef CAS.
- E. Weigold and I. E. McCarthy, Electron Momentum Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed.
- I. E. McCarthy, Can. J. Phys., 1996, 74, 703 CAS.
- I. E. McCarthy, Aust. J. Phys., 1998, 51, 593 Search PubMed.
- S. T. Hood, E. Weigold, I. E. McCarthy and P. J. O. Teubner, Nature Phys. Sci., 1973, 245, 63 Search PubMed.
- G. B. Hewitt, G. Cottrell, R. Northeast, S. Utteridge and M. J. Brunger, Meas. Sci. Technol., 2002 Search PubMed , manuscript in preparation.
- S. A. Canney, M. J. Brunger, I. E. McCarthy, P. J. Storer, S. Utteridge, M. Vos and E. Weigold, J. Electron Spectrosc. Relat. Phenom., 1997, 83, 65 CrossRef CAS.
- M. J. Brunger, I. E. McCarthy and E. Weigold, Phys. Rev. A, 1999, 59, 1245 CrossRef CAS.
- W. Adcock, M. J. Brunger, C. I. Clark, I. E. McCarthy, M. T. Michalewicz, W. von Niessen, E. Weigold and D. A. Winkler, J. Am. Chem. Soc., 1997, 119, 2896 CrossRef CAS.
- W. Adcock, M. J. Brunger, I. E. McCarthy, M. T. Michalewicz, W. von Niessen, F. Wang, E. Weigold and D. A. Winkler, J. Am. Chem. Soc., 2000, 122, 3892 CrossRef CAS.
- F. Wang, H. Mackenzie-Ross, D. A. Winkler, I. E. McCarthy, L. Campbell and M. J. Brunger, J. Comput. Chem., 2001, 22, 1321 CrossRef CAS.
- H. Mackenzie-Ross, M. J. Brunger, F. Wang, W. Adcock, N. Trout and D. A. Winkler, Chem. Phys. Lett., 2002 Search PubMed , manuscript in preparation.
- I. E. McCarthy and E. Weigold, Rep. Prog. Phys., 1988, 51, 299 CrossRef CAS.
- E. Harting and F. H. Read, Electrostatic Lenses, Elsevier, Amsterdam, 1976 Search PubMed.
- L. Frost and E. Weigold, J. Phys. B: At. Mol. Phys., 1982, 15, 2531 CrossRef CAS.
- P. R. Bevington and D. K. Robinson, Data Reduction and Error Analysis for the Physical Sciences, McGraw-Hill, New York, 1990 Search PubMed.
- M. Casida, Phys. Rev. A, 1995, 51, 2005 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- W. von Niessen, J. Schirmer and L. S. Cederbaum, Comput. Phys. Rep., 1984, 1, 58 Search PubMed.
- P. Duffy, D. P. Chong, M. Casida and D. R. Salahub, Phys. Rev. A, 1994, 50, 4707 CrossRef CAS.
- J. Andzelm and E. J. Wimmer, J. Chem. Phys., 1992, 96, 1290 CrossRef.
- A. Komornicki and G. J. Fitzgerald, J. Chem. Phys., 1993, 98, 1398 CrossRef CAS.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560 CAS.
- B. I. Dunlap, J. W. D. Connolly and J. R. Sabin, J. Chem. Phys., 1979, 71, 4993 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822 CrossRef.
- J. P. Perdew, in Electronic Properties of Solids 91, Akademie Verlag, Berlin, 1991 Search PubMed.
- J. P. Perdew, Phys. Rev. B, 1992, 46, 6671 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- M. Schmidt, K. K. Baldridge, J. A. Boatz, J. H. Jensen, S. Koseki, M. Gordon, K. A. Nguyen, T. L. Windurs and S. T. Elbert, QCPE Bull., 1984, 14, 52 Search PubMed.
- T. H. Dunning, J. Chem. Phys., 1971, 55, 716 CrossRef.
- K. Wiberg, J. Am. Chem. Soc., 1983, 105, 1227 CrossRef CAS.
- D. A. Winkler, M. T. Michalewicz, F. Wang and M. J. Brunger, J. Phys. B: At., Mol. Opt. Phys., 1999, 32, 3239 CrossRef CAS.
- C. E. Brion, in Correlations and Polarisation in Electronic and Atomic Collisions and (e,2e) Reactions, IOP Publishing, Bristol, 1992, p. 171 Search PubMed.
- M. T. Michalewicz, D. A. Winkler, M. J. Brunger, I. E. McCarthy and W. von Niessen, in Computational Chemistry and Chemical Engineering, World Scientific, Singapore, 1997, p. 280 Search PubMed.
- J. J. Neville, Y. Zheng, B. P. Hollebone, N. M. Cann, C. E. Brion, C.-K. Kim and S. Wolfe, Can. J. Phys., 1996, 74, 773 CAS.
- W. Adcock, M. J. Brunger, M. T. Michalewicz and D. A. Winkler, Aust. J. Phys., 1998, 51, 707 Search PubMed.
- C. A. Coulson and W. E. Duncanson, Proc. Cambridge. Philos. Soc., 1941, 37, 55 Search PubMed.
- C. A. Coulson and W. E. Duncanson, Proc. Cambridge. Philos. Soc., 1941, 37, 397 Search PubMed.
- C. A. Coulson and W. E. Duncanson, Proc. Cambridge. Philos. Soc., 1942, 38, 100 Search PubMed.
- C. A. Coulson and W. E. Duncanson, Proc. Cambridge. Philos. Soc., 1943, 39, 180 Search PubMed.
- I. R. Epstein, Acc. Chem. Res., 1973, 6, 175.
- I. R. Epstein and A. C. Tanner, in Compton Scattering, McGraw-Hill, New York, 1977, p. 209 Search PubMed.
- D. L. Cooper and N. L. Allan, in Molecular Similarity and Reactivity: From Quantum Chemical to Phenomenological Approaches, Kluwer Academic, Dordrecht, 1995, p. 31 Search PubMed.
- N. L. Allan and D. L. Cooper, Top. Curr. Chem., 1995, 173, 85 CAS.
- E. F. McCoy and M. J. Sykes, Chem. Phys. Lett., 1999, 313, 707 CrossRef CAS.
- E. R. Davidson, Can. J. Phys., 1996, 74, 757 CAS.
- P. Duffy, Can. J. Phys., 1996, 74, 763 CAS.
- A. Bawagan, C. E. Brion, E. R. Davidson and D. Feller, Chem. Phys., 1987, 113, 19 CrossRef CAS.
- E. Hirota and C. Matsumara, J. Chem. Phys., 1973, 59, 3038 CrossRef CAS.
- A. Maki and R. Toth, J. Mol. Spectrosc., 1965, 17, 136 CrossRef CAS.
- R. Hoffman, Acc. Chem. Res., 1971, 4, 1 CrossRef CAS.
- R. Hoffman, E. Heilbronner and R. Gleiter, J. Am. Chem. Soc., 1970, 92, 706 CrossRef.
- E. Heilbronner and A. Schmelzer, Helv. Chim. Acta, 1975, 58, 936 CrossRef CAS.
- S. W. Braidwood, M. J. Brunger, E. Weigold, W. von Niessen and V. G. Zakrzewski, J. Phys. B: At., Mol. Opt. Phys., 1994, 27, 2075 CrossRef CAS.
- C. Baker and D. W. Turner, Chem. Commun., 1969, 480 RSC.
- R. K. Thomas and H. Thompson, Proc. R. Soc. London, Ser. A, 1974, 339, 29 CAS.
- G. Bieri, F. Burger, E. Heilbronner and J. P. Maier, Helv. Chim. Acta, 1977, 60, 2213 CrossRef CAS.
- Z. Z. Yang, L. S. Wang, Y. T. Lee, D. A. Shirley, S. Y. Huang and W. A. Lester, Jr., Chem. Phys. Lett., 1990, 171, 9 CrossRef CAS.
- P. Baltzer, B. Wannberg, M. Lundqvist, L. Karlsson, D. M. P. Holland, M. A. MacDonald and W. von Niessen, Chem. Phys., 1995, 196, 551 CrossRef CAS.
- R. J. F. Nicholson, I. E. McCarthy and M. J. Brunger, Aust. J. Phys., 1998, 51, 691 Search PubMed.
- R. J. F. Nicholson, I. E. McCarthy and W. Weyrich, J. Phys. B: At., Mol. Opt. Phys., 1999, 32, 3873 CrossRef CAS.
- M. J. Brunger, D. A. Winkler, M. T. Michalewicz and E. Weigold, J. Chem. Phys., 1998, 108, 1859 CrossRef CAS.
- M. J. Brunger, S. W. Braidwood, I. E. McCarthy and E. Weigold, J. Phys. B: At., Mol. Opt. Phys., 1994, 27, L597 CrossRef CAS.
- K. B. Wiberg and F. H. Walker, J. Am. Chem. Soc., 1982, 104, 5239 CrossRef CAS.
- K. B. Wiberg, Acc. Chem. Res., 1984, 17, 379 CrossRef CAS.
- L. Hedberg and K. Hedberg, J. Am. Chem. Soc., 1985, 107, 7257 CrossRef CAS.
- K. B. Wiberg, W. P. Dailey, F. H. Walker, S. T. Waddell, L. S. Crocker and M. Newton, J. Am. Chem. Soc., 1985, 107, 7247 CrossRef CAS.
- K. B. Wiberg, R. E. Rosenberg and S. T. Waddell, J. Phys. Chem., 1992, 96, 8293 CrossRef CAS.
- K. B. Wiberg, R. F. W. Bader and C. D. H. Lau, J. Am. Chem. Soc., 1987, 109, 985 CrossRef CAS.
- O. Schafer, M. Allan, G. Szeimies and M. Sankjohanser, J. Am. Chem. Soc., 1992, 114, 8180 CrossRef CAS.
- M. D. Levin, P. Kaszynski and J. Michl, Chem. Rev., 2000, 100, 169 CrossRef CAS.
- E. Honegger, H. Huber, E. Heilbronner, W. P. Dailey and K. B. Wiberg, J. Am. Chem. Soc., 1985, 107, 7172 CrossRef CAS.
- W. Adcock, M. J. Brunger, C. I. Clark, I. E. McCarthy, E. Weigold, M. T. Michalewicz, D. A. Winkler and W. von Niessen, Chem. Phys. Lett., 1995, 244, 433 CrossRef CAS.
- T. S. Slee, in Modern Models of Bonding and Delocalisation, VCH Publishers, New York, 1988, p. 63 Search PubMed.
- K. B. Wiberg, R. F. W. Bader and C. D. H. Lau, J. Am. Chem. Soc., 1987, 109, 1001 CrossRef CAS.
- M. D. Newton and J. M. Schulman, J. Am. Chem. Soc., 1972, 94, 773 CrossRef CAS.
- R. M. Jarret and L. Cusumano, Tetrahedron Lett., 1990, 31, 171 CrossRef CAS.
- M. L. Herr, Tetrahedron, 1977, 33, 1897 CrossRef CAS.
- N. V. Riggs, V. Zoller, M. T. Nguyen and L. Radom, J. Am. Chem. Soc., 1992, 114, 4354 CrossRef CAS.
- J. E. Jackson and L. C. Allen, J. Am. Chem. Soc., 1984, 106, 591 CrossRef CAS.
- T. Kar and K. Jug, Chem. Phys. Lett., 1996, 256, 201 CrossRef CAS.
- P. E. Eaton and T. W. Cole, Jr., J. Am. Chem. Soc., 1964, 86, 962 CrossRef CAS.
- P. E. Eaton and T. W. Cole, Jr., J. Am. Chem. Soc., 1964, 86, 3157 CrossRef CAS.
- P. E. Eaton, Angew. Chem., Int. Ed. Engl., 1992, 31, 1421 CrossRef.
- J. Tsanaktsidis, Adv. Strain. Org. Chem., 1997, 6, 67 Search PubMed.
- T. Yildirim, P. M. Gehring, D. A. Neumann, P. E. Eaton and T. Emrick, Carbon, 1998, 36, 809 CrossRef CAS.
- E. Fleischer, J. Am. Chem. Soc., 1964, 86, 3889 CrossRef CAS.
- T. Yildirim, P. M. Gehring, D. A. Neumann, P. E. Eaton and T. Emrick, Phys. Rev. Lett., 1997, 78, 4938 Search PubMed.
- S. L. Richardson and J. L. Matins, Phys. Rev. B, 1998, 58, 15307 Search PubMed.
- L. Hedberg, K. Hedberg, P. E. Eaton, N. Nodari and A. G. Robiette, J. Am. Chem. Soc., 1991, 113, 1514 CrossRef CAS.
- W. Schubert, M. Yoshimine and J. J. Pacansky, J. Phys. Chem., 1981, 85, 1340 CrossRef CAS.
- J. T. Edward, P. G. Farrell and G. E. Langford, J. Am. Chem. Soc., 1976, 98, 3075 CrossRef CAS.
- E. W. Della, P. T. Hine and H. K. Patney, J. Org. Chem., 1977, 42, 2940 CrossRef CAS.
- C. P. Vlahacos, H. F. Hameka and J. O. Jensen, Chem. Phys. Lett., 1996, 259, 283 CrossRef CAS.
- E. W. Della, E. F. McCoy, H. K. Patney, G. L. Jones and F. A. Miller, J. Am. Chem. Soc., 1979, 101, 7441 CrossRef CAS.
- K. Miaskiewicz and D. A. Smith, Chem. Phys. Lett., 1997, 270, 376 CrossRef CAS.
- N. Bodor, M. J. S. Dewar and S. D. Worley, J. Am. Chem. Soc., 1970, 92, 19 CrossRef CAS.
- P. Bischof, P. E. Eaton, R. Gleiter, E. Heilbronner, T. B. Jones, H. Musso, A. Schmelzer and R. Stober, Helv. Chim. Acta, 1978, 61, 547 CrossRef CAS.
- J. M. Schulman, C. R. Fischer, P. Soloman and T. J. Venanzi, J. Am. Chem. Soc., 1978, 100, 2949 CrossRef CAS.
- J. Almlöf and T. Jonvik, Chem. Phys. Lett., 1982, 92, 267 CrossRef CAS.
- W. von Niessen, J. Schirmer and L. S. Cederbaum, Comput. Phys. Rep., 1984, 1, 59 Search PubMed.
- A. Almenningen, T. Jonvic, H. D. Martin and T. Urbanek, J. Mol. Struct., 1985, 128, 239 CrossRef CAS.
- E. Hirota, M. Fujitake, E. W. Della, P. E. Pigou and J. S. Chickos, J. Mol. Struct., 1988, 190, 235 CrossRef CAS.
- A. S. Pine, A. G. Maki, A. G. Robiette, B. J. Krohn, J. K. G. Watson and T. Urbanek, J. Am. Chem. Soc., 1984, 106, 891 CrossRef CAS.
- B. S. Jursic, THEOCHEM, 1997, 394, 15 CrossRef CAS.
- R. Hoffmann, A. Imamura and W. J. Hehre, J. Am. Chem. Soc., 1968, 90, 1499 CrossRef CAS.
- P. Bischof, J. A. Hashmall, E. Heilbronner and V. Harnung, Helv. Chim. Acta, 1969, 52, 1745 CrossRef CAS.
- G. Bieri, F. Burger, E. Heilbronner and J. P. Maier, Helv. Chim. Acta, 1977, 60, 2213 CrossRef CAS.
- W. von Niessen and G. H. F. Diercksen, J. Electron Spectrosc. Relat. Phenom., 1979, 16, 351 CrossRef CAS.
- E. Heilbronner and H. D. Martin, Helv. Chim. Acta, 1972, 55, 1490 CrossRef CAS.
- V. Galasso, Chem. Phys., 1989, 138, 231 CrossRef CAS.
- M. Takahashi, R. Ogino and Y. Udagawa, Chem. Phys. Lett., 1998, 288, 714 CrossRef CAS.
- H. Mackenzie-Ross, M. J. Brunger, F. Wang, W. Adcock, N. Trout, I. E. McCarthy and D. A. Winkler, J. Electron Spectrosc. Relat. Phenom., 2002 Search PubMed , in the press.
- H. Mackenzie-Ross, M. J. Brunger, F. Wang, W. Adcock, E. Weigold and D. A. Winkler, J. Am. Chem. Soc., 2002 Search PubMed , manuscript in preparation.
- G. Knuchel, G. Grassi, B. Vogelsanger and A. Bauder, J. Am. Chem. Soc., 1993, 115, 10845 CrossRef CAS.
- J. F. Chiang, R. Chiang, K. C. Lu, E.-M. Sung and M. D. Harmony, J. Mol. Struct., 1977, 41, 67 CrossRef CAS.
- M. N. Paddon-Row, S. S. Wong and K. D. Jordan, J. Am. Chem. Soc., 1990, 112, 1710 CrossRef CAS.
- C. R. Castro, R. Dutler, A. Rauk and H. Wieser, THEOCHEM, 1987, 152, 241 CrossRef.
- M. Z. Zgierski and F. Zerbetto, J. Chem. Phys., 1993, 98, 14 CrossRef CAS.
- R. Gleiter, H. Lange and O. Borzyk, J. Am. Chem. Soc., 1996, 118, 4889 CrossRef CAS.
- R. Gleiter, B. Gaa, C. Sigwart, H. Lange, O. Borzyk, F. Rominger, H. Irngartinger and T. Oeser, Eur. J. Org. Chem., 1998, 171 CrossRef CAS.
- A. Dorn, A. Elliot, X. Guo, J. M. Hurn, J. C. A. Lower, S. F. E. Mazevet, I. E. McCarthy, Y. Shen and E. Weigold, J. Phys. B: At., Mol. Opt. Phys., 1997, 30, 4097 CrossRef CAS.
- A. Dorn, A. Elliot, J. C. A. Lower, E. Weigold, J. Berakdar, H. Engelns and H. Klar, Phys. Rev. Lett., 1998, 80, 257 CrossRef CAS.
Footnote |
| † Low-resolution experiments refer to those with a coincidence energy resolution (spread) of about 1 eV, full-width-at-half-maximum (FWHM), or greater. |
| This journal is © The Royal Society of Chemistry 2002 |