Role of protolytic interactions in photo-aging processes of carminic acid and carminic lake in solution and painted layers
Gianna
Favaro
*,
Costanza
Miliani
,
Aldo
Romani
and
Manuela
Vagnini
Università di Perugia, Dipartimento di Chimica, 06123 Perugia, Italy. E-mail: favaro@phch.chm.unipg.it; Fax: 039 075 585 5598
First published on 7th December 2001
Abstract
In this paper absorption and fluorescence spectra and emission quantum yields and lifetimes of the red colorants, carminic acid and its metal complex, carminic lake, were studied in solution and on painted surfaces. Accelerated photo-aging of carminic acid and lake was investigated in solution, in the presence and absence of a binder (arabic gum) commonly used in water-colour painting, while natural ageing was followed for several months on water-colour painted paper.
The study of carminic acid in water as a function of pH showed that the absorption spectrum changes with pH. Four acid–base dissociation steps were detected and the corresponding pKs were determined from spectrophotometric and fluorimetric titrations. The fluorescence quantum yields (in the 10−2–10−4 range) and the lifetimes (on the sub-nanosecond timescale, 90–1000 ps) were markedly dependent on the pH of the medium. Excited state pK*s were calculated by means of the Förster cycle. The acidity decreased upon excitation for the first deprotonation step involving the –COOH group (ΔpK* = −1.9), but increased slightly for the successive deprotonation steps involving three phenolic hydroxy groups (ΔpK* = 0.6, 2.0 and 0.2, respectively).
The results obtained from the aging experiments indicate that both carminic acid and lake are bleached during irradiation. While the binder prevents lake from fading, it destabilises the carminic acid. These findings are discussed in the light of the interactions of the dye with the solvent and matrix.
Introduction
Lake pigments are translucent dyes prepared by precipitating or adsorbing an organic dye onto an inert inorganic substrate. These coloured substances were essential constituents of the artist's palette from the fourteenth to the nineteenth centuries.1 In this work, absorption and luminescence properties of carminic acid (CA) and its metal complex (carminic lake, CA–Al) were investigated in solution and on painted surfaces.Carminic acid is a β-C-glycopyranosyl derivative of anthraquinone (Fig. 1) which is extracted from the cochineal, a tropical American insect that feeds on certain species of cactus.
 | ||
| Fig. 1 Structural formula of carminic acid (CA). | ||
The related lake is an aluminium two-ligand complex, in which the Al atom co-ordinates two dye molecules through the 5-hydroxy oxygen and the adjacent carbonyl oxygen, forming a six-membered chelate ring structure.
This molecule belongs to the class of anthraquinonic dyes, which have been studied extensively due to their practical application as colorants (e.g., in paints, foods, cloth, etc.). In addition, there is basic interest in these molecules because modest structural changes induce drastic modifications in the absorption and fluorescence characteristics.2–4 The peculiar spectral behaviour of these compounds has been explained by considering the stability of intramolecular hydrogen-bonding in the ground and excited states, which may or may not undergo excited state intramolecular proton transfer (ESIPT) from the phenolic hydroxy group to the carbonyl oxygen upon excitation.5
Over the last decade, great interest has been devoted to carminic acid, mainly in relation to its extensive use as a natural food and cosmetic pigment. Various studies have investigated photodegradation,6 fluorescence properties in both aqueous solution7 and organic solvents,8 reorientation dynamics,8 self-assembly,9 and acid–base behaviour. 6,10
In the present paper, attention is focused on the use of carminic acid and carminic lake in paints and the possible colour changes or colour fading that they may undergo with aging.
Paintings are exposed to atmospheric pollutants and solar irradiation, and undergo natural aging. This exposure may cause deterioration, particularly by changing the original colours. It is important to know which chromatic changes occur as a result of environmental changes. Acid rain is one form of pollution that attacks painted artefacts. By studying protolytic equilibria involving the multiple acidic centres of this molecule and the accompanying colour changes, information can be obtained about the environmental effects as well as about possible interactions of the dye with the binders and varnishes present in the painted layers. Even though several papers from the literature report results on acid-base titration and fluorescence measurements of carminic acid,6–10 some discrepancies between the literature data and our findings induced us to reproduce and present both spectral and titration data in this paper.
Light can also alter colours and damage painted surfaces. Light-induced effects may have dramatic consequences on the chromatic features of paintings. In order to evaluate the aging that might occur indoors in a museum, painted layers and dye solutions (for the purpose of comparison) were irradiated with a lamp to simulate daylight. Their spectral changes were periodically analysed.
Experimental
Materials
The carminic acid was purchased from Aldrich and used without further purification. The carminic lake was purchased from Zecchi (Florence, Italy). The organic solvent used, DMSO, was a Fluka spectrograde product. Arabic gum was from Winsor & Newton, London. To determine the pK values, Britton buffers were used in the 2–12 pH range; diluted HClO4 and NaOH solutions were used in the acidic and alkaline regions, respectively. Re-distilled water was used to prepare the buffers.To prepare the samples of painted surfaces, the dye was mixed with the binder and painted onto a piece of paper.
Apparatus
The absorption spectra in solution were recorded with a Perkin Elmer Lambda 16 spectrophotometer. The Perkin Elmer accessory for reflectance measurements was used to record absorption on the painted surfaces.Corrected emission spectra were obtained using a Spex Fluorolog-2 FL 112 spectrofluorimeter controlled by Spex DM3000F spectroscopy software.
To record temperature and solar radiation values during the natural aging experiments, a data-logger instrument for recording and statistically elaborating temperature and radiance data (Babuc, from LSI, Milan, Italy) was used. Typical histograms of solar radiance and temperature distribution during the period of observation (eight months) are reported in Fig. 2.
 | ||
| Fig. 2 Radiation intensity (a) and temperature (b) distributions during an eight-month period. | ||
For the accelerated photo-aging trials, carried out on the dye in DMSO solution, a 150 W Xenon lamp filtered through a Pyrex filter was used. The exposure times did not exceed 50 hours.
Measurement conditions
The emission quantum yields (ΦF) were obtained by comparing corrected areas of the sample (areaS) and the standard (areaST) emissions, using quinine sulfate in 0.5 M H2SO4, ΦST = 0.546, as the standard.11 The formula in eqn. (1)| ΦF = ΦST × (AST/AS) × (areaS/areaST) × (nS2/nST2) | (1) |
Sample concentrations were adjusted in order to keep the absorbance within 0.2–0.5. This is a compromise between needing to have a detectable signal and the requirement to be within the linear range of the absorbance. The relatively large Stokes shifts militate against self-absorption of the fluorescence emission. The solvent contribution to the emission signal was subtracted, when necessary. The accuracy in the ΦF values is estimated to be within 10% for ΦF ≥ 10−3 and within 20% for ΦF < 10−3.
The emission lifetimes (τF) were determined by using the Pockel's cell of the Spex spectrofluorimeter accessory, which is based on the phase-shift method (time resolution ca. 20 ps). The light scattered by an aqueous glycogen solution (undetectable fluorescence) was used as the standard. The accuracy of the data obtained depended on the intensity of the emission. The data were processed using Global Unlimited™ 3.0 software, which allows the analysis of multiple decays of up to four components.
Spectrophotometric and fluorimetric titrations
Low ionic strength Britton buffers (μ = 0.01 mol dm−3) were used to determine the pKs. The pH values were measured with an Orion 9103 pH-meter.The dissociation constants were obtained from the inflection points in the absorbance vs. pH curves at the wavelengths of the maximum difference in absorbance of the equilibrating species. In the fluorimetric titrations, the excitation was carried out at an isosbestic point between the spectra of the equilibrating species; the inflection points of the emission intensity vs. pH curves at suitable wavelengths gave the dissociation constant values. Sample concentrations were on the order of 10−5 mol dm−3. Given the low concentration and low ionic strength of the buffer solutions, the activity coefficient ratio of the acid–base couples was considered to be one.
Results and discussion
Effect of pH on the spectral characteristics of carminic acid
The effect of pH on the absorption and emission spectra of carminic acid was investigated in the 0.9–13 pH range. Dilute solutions (<5 × 10−5 mol dm−3) were used in order to avoid aggregation. The lake could not be investigated in aqueous solution due to its poor solubility.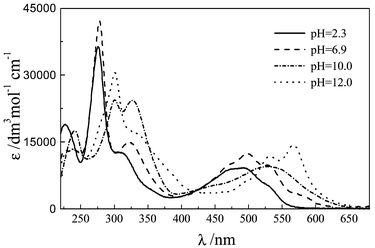 | ||
| Fig. 3 Absorption spectra of CA in aqueous solution at various pH values. | ||
 | ||
| Fig. 4 Fluorescence spectra (solid line) normalised to absorption spectra (dotted line) of the four species spectrally distinguishable when the pH changes. | ||
The fluorescence lifetimes, determined by the phase-shift method, are of the order of hundreds of picoseconds. At certain pH values, the decay kinetics fit a bi-exponential function well; contributions from the two species are pH dependent. Detection of two distinct lifetimes is a good indication that acidic and basic excited species do not equilibrate within the excited state lifetime. The rate parameters for emission, kF = ΦF/τ, and radiationless deactivation, kNR = (1/τ) − kF, were determined from the experimental ΦF and τ data and are reported in Table 1.
| pH (status) | Φ F | τ/ps | k F/107 s−1 | k NR/109 s−1 |
|---|---|---|---|---|
| a Data taken from ref. 7. | ||||
| 0.9 (Neutral) | 1.0 × 10−3 (1.33 × 10−3)a | (93)a | (1.4)a | (11.0)a |
| 2.3 | 1.8 × 10−3 | 94 | 1.9 | 10.6 |
| 3.3 (Mono-anion) | 3.3 × 10−3 | |||
| 6.9 (Di-anion) | 1.3 × 10−2 | 940 | 1.4 | 1.0 |
| 10.0 (Tri-anion) | 6.2 × 10−4 | Not measurable | ||
| 12.4 (Tetra-anion) | 2.5 × 10−3 | 330 | 0.75 | 2.9 |
The ΦF and τ values were strongly pH dependent. They were in the 10−2–10−4 and 90–1000 ps ranges, respectively. The kF values were of the order of 107 s−1 over the entire pH range explored, while kNR varied from 1 × 1010 to 10 × 1010 s−1 depending on the pH value. The kNR rate constant includes both internal conversion to the ground state and intersystem crossing to the triplet state. Triplet–triplet absorption was, in fact, detected in a butanol solution by laser flash-photolysis (λexc = 470 nm). The transient produced, with a lifetime on the microsecond timescale (τ = 3 μs), was quenched by oxygen at a diffusion-controlled rate.
Acid–base properties
To determine the pKs of the protolytic dissociations, spectrophotometric and fluorimetric titrations were employed. Inflection points were observed within the pH intervals where isosbestic points were maintained between the two equilibrating species.As for the 2-hydroxyanthraquinones,12 the fluorimetric titration curves showed inflection points at the same pH values as the spectrophotometric ones, for pK1, pK2 and pK3. Therefore, these curves also provided the ground state pKs, since the protolytic equilibrium in the excited state was not established within the lifetime of that state.13 The deprotonation step occurring in the acidic range (pK0 = 2.9), which was only detected by fluorescence, could have been assigned to excited state acid–base equilibrium. However, comparison with literature values6,10 indicates that pK0 corresponds to the deprotonation of the most acidic site in the ground state molecule. The pKs determined are reported in Table 2 and are compared with values from the literature;6,9,10 the fourth pK (∼13) was only roughly estimated.9
The excited state pKs*, calculated by use of the thermodynamic Förster cycle,14 are also reported. The frequency differences between 0–0 levels, needed to calculate the pK*, were obtained from the intersections of the normalised absorption and fluorescence spectra. The acidity may increase or decrease upon excitation. For carminic acid, the acidity decreased in the excited state for the first deprotonation, while, for the successive deprotonations, a slight increase in acidity was observed upon excitation, even though the increase was much smaller than for other hydroxyanthraquinones.12,15
These results give a complete view of the acid–base equilibria of this molecule and allow the spectra to be assigned to the molecular species that interconvert one into the other as the pH changes. In carminic acid, where several acidic centres are present, the first dissociation step (pK0 = 2.9) definitely occurs at the carboxylic group in the 2 position, which is the most acidic site. It is more acidic than 2-anthraquinoic acid (pK = 4.2),16 due to the inductive electron-attracting effect of the phenolic OH group in the 3 position, which decreases the charge density in the 2 position and consequently lowers the pK of the COOH group. Deprotonation at the carboxylic group does not produce appreciable spectral changes in this molecule, where the chromatic properties are essentially determined by the hydroxyanthraquinone chromophore. Further support for this assignment is the pK increase in the excited state, which generally decreases for phenolic hydroxy groups. Accordingly, the acidity increases in the analogous 2-anthraquinoic acid upon excitation (ΔpK* ∼ −2).16
The following acid–base equilibration (pK1 = 5.4) involves the 6-hydroxy group since the 5- and 8-OH groups are less acidic due to their position with respect to the carbonyl, which reduces the hydrogen mobility through hydrogen bonding. Moreover, the 6-hydroxy group is far enough away from the negatively charged site, COO−, that its ionisation is only weakly influenced by the repulsive electrostatic potential. This assignment is in contrast with that reported in the literature, where the pK = 5.62 is attributed to dissociation of the OH in the 5 position.10
Even though the double negative charge of the di-anion should lower the acidity of the other hydroxy groups, two further deprotonation steps (pK2 = 8.7 and pK3 = 12.2) were observed upon decreasing the acidity, assigned to the dissociation of the 3- and 8-hydroxy groups. The absorption spectrum of the tetra-anion, which is stable at pH > 12, is structurally similar to that of the purpurin di-anion, which involves 2- and 4-hydroxy deprotonations.12 Since the 2 and 4 positions in purpurin are equivalent to the 6 and 8 positions in carminic acid, the latter are probably both deprotonated at pH = 12.
The photophysical data reported in Table 2 indicate that the variations in ΦF and τF with deprotonation essentially depend on the different contributions of non-radiative relaxation, since all the kF values are of the same order of magnitude. The relatively long lifetime of carminic acid at pH = 6 (di-anionic form) is the result of the intramolecular hydrogen bonds of the 5- and 8-hydroxy groups that keep the ion rigid. The clear-cut decrease of emission intensity of the tri-anion (stable at pH ∼ 10) favours the deprotonation at the 8 position. The further increase of the emission quantum yield of the tetra-anionic form is probably related to the loss of relaxation paths through OH vibrations. For this molecule, acid–base equilibria in the excited state involve intramolecular hydrogen-bonded forms, as in the ground state, due to minute changes in the geometry upon excitation. Semiempirical calculations have shown that there are no marked changes in dipole moment upon excitation.17 This also explains the small increase in acidity upon excitation.
Effects of environment and aging on the spectral properties of CA and CA–Al
An important aspect of studies on the effect of pH on colorants used in artistic painting is its potential to interpret the interactions of colorants with atmospheric pollutants, as well as within their own chemical environment. In painting, dyes and pigments are usually dispersed in a binder and protected by a top layer of transparent varnish. The spectral features of the absorption and emission bands of the colorant depend on both the aging and interaction of the dye with the additives. Thus, spectra can be used as a diagnostic test of the medium used by the artist and can help to evaluate how the durability of the colorant can be increased or decreased by the presence of additives.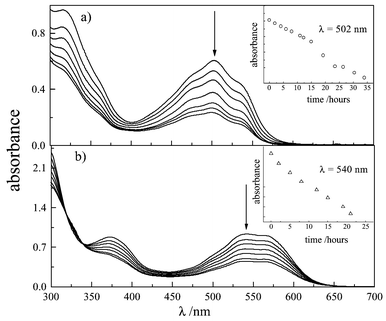 | ||
| Fig. 5 Effect of irradiation on the absorption spectrum of DMSO solutions of carminic acid (7.7 × 10−5 mol dm−3) in the absence (a) and in the presence (b) of arabic gum (1 ∶ 100 v/v). Inserts: bleaching kinetics followed at constant wavelength. | ||
 | ||
| Fig. 6 Effect of irradiation on the absorption spectrum of DMSO solutions of carminic lake (2.8 × 10−5 mol dm−3) in the absence (a) and in the presence (b) of arabic gum (1 ∶ 100 v/v). Inserts: bleaching kinetics followed at constant wavelength. | ||
When arabic gum, usually used as a binder with hydro-soluble dyes, was added to the solutions, the CA spectrum changed further [Fig. 5(b)], while that of CA–Al remained substantially unchanged [Fig. 6(b)]. The red-shift (λmax ≈ 550 nm) exhibited by the CA spectrum in the presence of the binder suggests that a further deprotonation occurs. The spectrum is, in fact, very close to that observed in aqueous solution at pH ≈ 10. Upon irradiation, the solution containing CA and arabic gum showed a very marked decoloration [Fig. 5(b)], while that containing the lake was highly photostable [Fig. 6(b)]. Thus, it has been established that arabic gum has a photo-stabilising effect on lake, by reducing the degradation compared with a solution of lake alone. Photobleaching quantum yields were estimated relative to the most photodegradable system, that is, CA + binder (Φ = 1) by using the relationship given in eqn. (2):
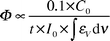 | (2) |
| CA | CA–Al | CA–arabic gum | CA–Al–arabic gum | |
|---|---|---|---|---|
| t ½ (hours) | 25 | 38 | 18 | 220 |
| Φ rel | 0.873 | 0.339 | 1 | 0.057 |
A study on the photodegradation of CA as a function of pH, showed that photodegradation increases as the pH increases.6 This is in agreement with the above measurements, since the greatest degradation occurred under conditions where the absorption spectrum showed the presence of a poly-deprotonated form, that is, for the CA-arabic gum system.
 | ||
| Fig. 7 Effect of irradiation on the reflectance spectrum of a water-painted paper with carminic acid. A′ = log 1/R (R is the reflectance signal). | ||
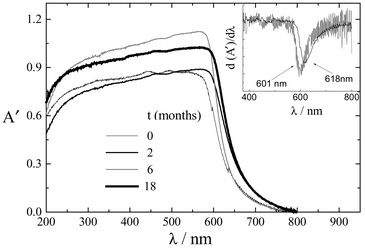 | ||
| Fig. 8 Effect of irradiation on the reflectance spectrum of water-painted paper with carminic lake. A′ = log 1/R (R is the reflectance signal). | ||
The reflectance spectra are less informative than normal absorption spectra. In this case, the inflection point, obtained from the first derivative (see inserts), is meaningful. Red shifts were observed for both the CA and CA–Al spectra with aging. The amount of degradation was estimated from the shift to the red of the minimum of the derivative. It can be seen from the figures that, during the same aging time (eighteen months), the spectrum of the CA painted paper red-shifted 43 nm, whereas that painted with lake shifted only 17 nm. This confirms that the aluminium complex is the most stable colorant, as indicated from the study in solution.
Conclusions
A study of the pH effect on absorption and fluorescence spectra of carminic acid allowed the ground and excited state pKs to be determined. In the pH range explored, four acid–base dissociation steps were detected. By assigning the absorption and fluorescence spectra, the different forms, stable at certain pH values, were characterised (neutral form, mono-anion, di-anion, tri-anion and tetra-anion). This provides a valuable analytical tool for investigating interactions involving the hydrogen transfer processes between the dye and its chemical environment.Carminic acid behaves like an acidichromic molecule since its aqueous solutions change colour as the pH changes. Emission is less sensitive to changes in the pH than absorption, since the fluorescence spectra of the differently protonated forms widely overlap. The fluorescence quantum yields and lifetimes are markedly dependent on the pH of the medium. Radiationless processes (internal conversion and intersystem crossing to the triplet) contribute greatly to excited singlet deactivation.
In the excited state, the neutral molecule becomes a weaker acid upon excitation, since the carboxy group is involved in the first dissociation step, while the hydroxy species, involved in the successive dissociation steps, are slightly stronger acids in the excited state. However, the pK decrease upon excitation is not as great as in other hydroxyanthraquinones.
The information about the spectral properties and their pH dependence has shed light on the spectral changes occurring in the presence of the binder. The absorption spectrum of the dye, before and after irradiation, depends on its environment. Thus, colour can be a diagnostic test of the medium used by the artist. All solutions were bleached upon irradiation and the degree of bleaching depended on the environment. The photoproducts absorb mainly in the UV region, thus photodegradation causes colour fading, not colour changes. When binder (arabic gum) was added to the solution, different effects were produced on the CA and CA–Al spectra and their photobleaching; the durability of CA decreased, while lake was efficiently stabilised. This explains why lakes have been widely used as colorants in painting. Finally, the spectral changes induced by photo-aging on painted surfaces may indicate the transformations that might occur indoors in a museum and may be used to interpret this effect in terms of the interactions of the dye with its environment.
Acknowledgements
This research was funded by the Ministero per l'Università e la Ricerca Scientifica e Tecnologica (Rome) and the University of Perugia in the framework of a “Progetto di Ateneo”. A grant from the Italian Consiglio Nazionale delle Ricerche is also acknowledged.References
- D. Saunders and J. Kirby, Natl. Gallery Techn. Bull., 1994, 15, 79 Search PubMed.
- A. Navas Diaz, J. Photochem. Photobiol. A, 1990, 53, 141 Search PubMed and references therein..
- K. Gollnick, S. Held, D. O. Màrtire and S. E. Braslavsky, J. Photochem. Photobiol., 1992, 69, 155 Search PubMed.
- C. Miliani, A. Romani and G. Favaro, Spectrochim. Acta, Part A, 1998, 54, 581 Search PubMed.
- D. K. Palit, H. Pal, T. Mukherjee and J. P. Mittal, J. Chem. Soc., Faraday Trans., 1990, 86, 3861 RSC and references therein; P. F. Barbara, P. K. Walsh and L. E. Brus, J. Phys. Chem., 1988, 93, 29 Search PubMed; M. H. van Benthem and G. D. Gillispie, J. Phys. Chem., 1984, 88, 2954.
- K. Jørgensen and L. H. Skibsted, Food Chem., 1991, 40, 25 CrossRef.
- H. Stapelfeldt, H. Jun and L. H. Skibsted, Food Chem., 1993, 48, 1 CrossRef CAS.
- J. P. Rasimas and G. J. Blanchard, J. Phys. Chem., 1995, 99, 11333 CrossRef CAS.
- J. P. Rasimas, K. A. Berglund and G. J. Blanchard, J. Phys. Chem., 1996, 100, 7220 CrossRef CAS.
- M. J. Schwing-Weill and S. Wechsler, Analusis, 1986, 14, 290 Search PubMed.
- S. R Meech and D. Phillips, J. Photochem., 1983, 23, 193 Search PubMed.
- C. Miliani, A. Romani and G. Favaro, J. Phys. Org. Chem., 2000, 13, 141 CrossRef CAS.
- J. F. Ireland and P. A. H. Wyatt, Adv. Phys. Org. Chem., 1976, 12, 131 Search PubMed.
- T. Förster, Z. Electrochem., 1950, 54, 531 Search PubMed.
- H. H. Richtol and B. R. Fitch, Anal. Chem., 1974, 46, 1749 CrossRef CAS.
- E. Vander Donckt, in Eléments de Photochimie Avancée, ed. P. Courtot, Hermann, Paris, France, 1972, p. 79 Search PubMed.
- J. P. Rasimas and G. J. Blanchard, J. Phys. Chem., 1994, 98, 12949 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |