A chemoenzymatic synthesis of the 12-membered macrolide (−)-cladospolide A
Martin G.
Banwell
*,
Katrina A.
Jolliffe
,
David T. J.
Loong
,
Kenneth J.
McRae
and
Filisaty
Vounatsos
Research School of Chemistry, Institute of Advanced Studies, The Australian National University, Canberra, ACT 0200, Australia. E-mail: mgb@rsc.anu.edu.au
First published on 3rd December 2001
Abstract
The enantiomerically pure cis-1,2-dihydrocatechol 7, which is obtained by microbial oxidation of chlorobenzene, has been converted, via a sequence of reactions including ring-closing metathesis and Yamaguchi lactonisation steps, into the natural product (−)-cladospolide A (1).
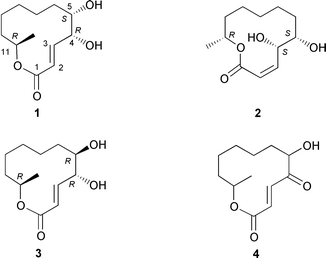
Cladospolides A (1)1 and B (2)2 were first isolated from the culture filtrate of the fungus Cladosporium cladosporioides FI-113 by Isogai and co-workers. Subsequently Fukuda et al. reported3 the isolation of cladospolide C (3), as well as congeners 1 and 2, from Cladosporium tenuissimum. Very recently, Omura et al. detailed4 the extraction of cladospolide D (4, stereochemistry undefined) from the fermentation broth of Cladosporium sp. FT-0012. Macrolide 1 has been shown to inhibit root growth in lettuce seedlings, whilst isomer 2 displays the opposite effect.2 Interestingly, compound 2 also appears to inhibit gibberellin biosynthesis as evidenced by its capacity to constrain shoot elongation of rice seedlings (Oryza sativa L.) without causing necrosis.3 Such interesting plant growth regulating properties have prompted several synthetic studies although these have all been focussed on compound 1. The first total synthesis of (−)-cladospolide A was described in 19875 by Mori and Maemoto who employed ethyl (R)-3-hydroxybutyrate as starting material and the stereogenic centre associated with this synthon was parlayed into C-11 of the target 1. The hydroxy groups at C-4 and C-5 were introduced via a Katsuki–Sharpless asymmetric epoxidation reaction while the macrolide ring of (−)-cladospolide A (1) was established using the Yamaguchi macrolactonisation procedure. A closely related synthesis was reported in the same year by Ichimoto et al.6 The third and most recent synthesis of (−)-cladospolide A was detailed in 1994 by Solladié and Almario7 who established all the stereogenic centers associated with the target via diastereoselective reduction of enantiomerically pure β-ketosulfoxides. Once again, the macrocyclic ring within target 1 was constructed via a late-stage Yamaguchi lactonisation step.
We now report a rather efficient synthesis of (−)-cladospolide A that employs, inter alia, three enzyme-mediated transformations and a ring-closing metathesis (RCM)8 step. The original plan, shown in retrosynthetic form in Fig. 1, was predicated on our recent success in using related chemistry to prepare the 18-membered macrolide (+)-aspicilin.9 Thus, we envisaged the 12-membered lactone ring of target 1 could be created via RCM of the terminal double-bonds within triene 5.8 Hydrogenation of the resulting olefin followed by removal of the TBDMS-protecting groups was then expected to deliver target 1. Sub-target 5 should be accessible via an E-selective Wadsworth–Horner–Emmons (WHE)-type coupling of aldehyde 6 with phosphonoacetate 8. The seven-carbon building block 6 was to be constructed from cis-1,2-dihydrocatechol 7, a material available in large quantity and enantiomerically pure form via toluene dioxygenase-mediated dihydroxylation of chlorobenzene.10 On the other hand, compound 8 was to be prepared by transesterification11 of readily available trimethyl phosphonoacetate with (R)-(−)-pent-4-en-2-ol [(−)-(9)].
 | ||
| Fig. 1 | ||
The route used to assemble compound 6 is shown in Scheme 1 and follows from earlier work.9,12 Thus, diol 7 was selectively reduced, with hydrogen in the presence of rhodium on alumina,12 to the dihydro analogue 1013 (75%), which was, in turn, converted under standard conditions into the corresponding bis-TBDMS-ether 11† {95%, [α]D −13 (c 7.4)‡}. When ozonolytic cleavage of alkene 11 was carried out in methanol at −78 °C and the ensuing peroxidic material subjected to reductive work-up with sodium borohydride, then the ester aldehyde 12 was obtained. The unstable nature of this last compound meant that it was best to effect its rapid methylenation, which was achieved with the Wittig reagent at 0 °C, and in this manner the unsaturated ester 13 {[α]D −9.1 (c 0.3)} was produced. This two step sequence was readily carried out on a multi-gram scale and compound 13 obtained in 65% overall yield from alkene 11. Direct reduction of ester 13 to the target, but unstable, aldehyde 6 could be achieved by reaction of the former compound with essentially equimolar quantities of diisobutylaluminium hydride (DIBAL-H) in hexane at −78 °C.
 | ||
Scheme 1
Reagents and conditions: (i) H2 (1 atm), 5% Rh on alumina, EtOH, 18 °C, 16 h; (ii) TBDMS-Cl (3.5 mol equiv.), imidazole (12.4 mol equiv.), DMF, 18 °C, 24 h; (iii) O3, MeOH, −78 °C, 0.5 h then NaBH4 (2.0 mol equiv.), 0 °C, 1 h; (iv) Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (1.5 mol equiv.), THF, 0–18 °C, 6.5 h; (v) DIBAL-H (1.1 mol equiv.), hexane, −78 °C, 0.75 h. CH2 (1.5 mol equiv.), THF, 0–18 °C, 6.5 h; (v) DIBAL-H (1.1 mol equiv.), hexane, −78 °C, 0.75 h. | ||
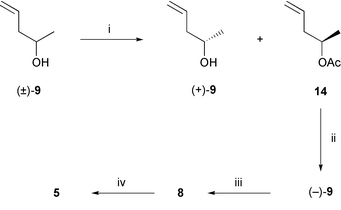
As a necessary prelude to synthesizing phosphonoacetate 8, access to the enantiomerically pure alcohol (−)-914 was required. In our hands the most effective route to gram quantities of this material involved initial reaction of the commercially available racemate (±)-9 with isopropenyl acetate in the presence of Candida antarctica lipase B15 (CALB, ex Novo Nordisk). Under appropriate conditions (Scheme 2) a chromatographically separable mixture of (+)-(S)-9 (60% yield, 99% ee§) and acetate 14 (80% yield, ca. 75% ee) was obtained. Treatment of the latter compound with CALB in phosphate buffer (pH 7.6) then provided enantiomerically pure samples of the target alcohol (−)-(R)-9 (60% yield).§ Reaction of alcohol (−)-9 with equimolar amounts of trimethyl phosphonoacetate and an excess of 4-(N,N-dimethylamino)pyridine (DMAP) in refluxing toluene for 6.5 h resulted in a smooth transesterification reaction11 and formation of the target diester 8 {[α]D +79 (c 1.1)} in ca. 75% yield. WHE-type coupling of this latter compound with aldehyde 6 then afforded the target RCM substrate 5 {65% ex compound 13, [α]D −3 (c 0.7)}. However, contrary to expectations,9 when this substrate was treated with either the first16 or the second17 generation Grubbs' catalyst the desired 12-membered macrocycle 15 (Scheme 3) was not formed. Rather, varying yields (90–100%) of an alternate RCM product, viz. cyclohexene 16, {[α]D −119 (c 0.5)} were observed.
 | ||
| Scheme 2 Reagents and conditions: (i) CALB (10% by weight), isopropenyl acetate (solvent), 25 °C, 1 h; (ii) CALB (10% by weight), aqueous phosphate buffer (pH 7.6), 25 °C, 16 h; (iii) (MeO)2P(O)CH2CO2Me (1 mol equiv.), DMAP (12 mol equiv.), toluene, 111 °C, 6.5 h; (iv) NaH (1.0 mol equiv.), THF, 0–18 °C, 2 h then compound 6 (0.83 mol equiv.), 0–18 °C, 1.5 h. | ||
ruthenium(iv) dichloride (18 mol%), CH2Cl2, 18 °C, 24 h.](/image/article/2002/P1/b110018a/b110018a-s3.gif) | ||
| Scheme 3 Reagents and conditions: (i) tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene](benzylidene)ruthenium(IV) dichloride (18 mol%), CH2Cl2, 18 °C, 24 h. | ||
On the basis of the foregoing observations it seemed clear that if a RCM-based strategy was to be employed in accessing the target macrolide 1 then the substrate to be used for such purposes should include just two C–C double bonds such that only one reaction pathway would be followed. To this end, various methods for temporarily masking the conjugated double bond within substrate 5 were pursued. However, all efforts along these lines proved fruitless, thus highlighting the need for a new approach to target 1. The revised strategy that followed is shown in Scheme 4 and involved initial conversion of ester 13 into the corresponding acid (64%) which was immediately subject to Yonemitsu esterification18 using alcohol (−)-9. The resulting ester 17 {100%, [α]D −5 (c 0.8)} readily engaged in an RCM reaction upon exposure to Grubbs' second generation catalyst17 so as to afford the ten-membered lactone 18 (73% at 75% conversion) as a single geometric isomer for which the (Z)-configuration is tentatively assigned.19 Hydrogenation of the latter compound and DIBAL-H reduction of the ensuing saturated lactone {89%, [α]D −2 (c 0.8)} then afforded the lactol 19 which reacted smoothly with the sodium salt of trimethyl phosphonoacetate to give the α,β-unsaturated ester 20 {43% from saturated lactone, [α]D −3 (c 0.4)}. Saponification of the last compound followed by acidification afforded the free acid 21 which was subject to a standard Yamaguchi lactonisation protocol20 thereby providing the bis-TBDMS-ether 22 (89% from 20) of (−)-cladospolide A (1). Initial attempts to effect desilylation of compound 22 involved the use of TBAF but only a complex mixture of products, some of which may arise via a retro-aldol process, was observed. After extensive experimentation with a wide range of fluoride ion sources, it was established that zinc tetrafluoroborate in water–acetonitrile21 could circumvent such difficulties and (+)-cladospolide A (1) (73%) was obtained as a crystalline solid, mp 90–91 °C (lit.1 mp 92–93 °C). The various spectroscopic data, including specific rotation {[α]D −53 (c 0.2); lit.5 [α]D −49 (c 0.23 in CHCl3)}, derived from the synthetic material were in agreement with those reported5 previously.
ruthenium(iv) dichloride (15 mol%), CH2Cl2, 18 °C, 24 h; (iii) H2 (1 atm), 10% Pd on C, EtOH, 18 °C, 6 h; (iv) DIBAL-H (2.0 mol equiv.), toluene, −78 °C, 10 min; (v) (MeO)2P(O)CH2CO2Me, NaH (0.9 mol equiv.), THF, 0–18 °C, 3 h then compound 19 (0.5 mol equiv.), 0–18 °C, 1 h; (vi) NaOH (3.7 mol equiv.), 4 ∶
1 v/v EtOH–H2O, 18 °C, 18 h then 1 M aq. HCl; (vii) 2,4,6-trichlorobenzoyl chloride (1.1 mol equiv.), Et3N (1.2 mol equiv.), THF, 18 °C, 2 h then DMAP (4.0 mol equiv.), toluene, 111 °C, 1 h; (viii) Zn(BF4)2 (5.0 mol equiv. of a 5 M aq. solution), acetonitrile, 18 °C, 24 h.](/image/article/2002/P1/b110018a/b110018a-s4.gif) | ||
| Scheme 4 Reagents and conditions: (i) NaOH (3.7 mol equiv.), 9 ∶ 1 v/v EtOH–H2O, 60 °C, 2 h then 1 M aq. HCl then 2,4,6-trichlorobenzoyl chloride (0.95 mol equiv.), (−)-9 (1.1 mol equiv.), Et3N (5.0 mol equiv.), DMAP (1.0 mol equiv.), toluene, 18 °C, 1 h; (ii) tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene](benzylidene)ruthenium(IV) dichloride (15 mol%), CH2Cl2, 18 °C, 24 h; (iii) H2 (1 atm), 10% Pd on C, EtOH, 18 °C, 6 h; (iv) DIBAL-H (2.0 mol equiv.), toluene, −78 °C, 10 min; (v) (MeO)2P(O)CH2CO2Me, NaH (0.9 mol equiv.), THF, 0–18 °C, 3 h then compound 19 (0.5 mol equiv.), 0–18 °C, 1 h; (vi) NaOH (3.7 mol equiv.), 4 ∶ 1 v/v EtOH–H2O, 18 °C, 18 h then 1 M aq. HCl; (vii) 2,4,6-trichlorobenzoyl chloride (1.1 mol equiv.), Et3N (1.2 mol equiv.), THF, 18 °C, 2 h then DMAP (4.0 mol equiv.), toluene, 111 °C, 1 h; (viii) Zn(BF4)2 (5.0 mol equiv. of a 5 M aq. solution), acetonitrile, 18 °C, 24 h. | ||
In summary, a concise synthesis of the title natural product has been achieved via a tandem RCM–Yamaguchi lactonisation sequence employing the enzymatically derived synthons 7 and (−)-9. The strategies and tactics reported herein should be applicable to the preparation of a range of related macrolides, especially compounds 2, 3 and 4, and studies directed toward this end are now underway in these laboratories.
Experimental
Compound 18
A magnetically stirred solution of diene 17 (125 mg, 0.27 mmol) in degassed CH2Cl2 (140 mL) and maintained under a nitrogen atmosphere was treated, via cannula, with a solution of tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene](benzylidene)ruthenium(IV) dichloride17 (35 mg, 0.04 mmol, 15 mol%, ex Strem Chemicals Inc.) in CH2Cl2 (10 mL). The resulting mixture was stirred at 18 °C for 24 h then (HOCH2)3P22 (∼50 mg) and Merck silica gel 60 (∼200 mg) were added to the reaction mixture. The resulting slurry was stirred at 18 °C for 1 h then filtered through a ca. 3 cm deep pad of Merck silica gel 60 which was washed with CH2Cl2 (ca. 10 mL). The combined filtrates were concentrated under reduced pressure and the light-yellow oil thus obtained was subject to flash chromatography (silica gel, 98 ∶ 2 v/v hexane–Et2O elution). In this manner two fractions, A (Rf 0.30 in 95 ∶ 5 v/v hexane–EtOAc) and B (Rf 0.28 in 95 ∶ 5 v/v hexane–EtOAc), were obtained.Concentration of fraction A gave the starting diene 17 (32 mg, 25% recovery) which was identical, in all respects, with an authentic sample.
Concentration of fraction B gave the title compound18 (61 mg, 73% yield at 75% conversion) as clear colourless oil, [α]D +0.8 (c 0.7). νmax (KBr) 2931, 2857, 1729, 1465, 1255, 1155, 1073, 957, 837 and 777 cm−1; δH (300 MHz, CDCl3) 5.45–5.35 (2H, complex m), 5.10 (1H, m), 4.28 (1H, br s), 3.98 (1H, dd, J 10.9 and 4.7 Hz), 2.72 (1H, br m), 2.40 (1H, br m), 2.06–1.76 (3H, complex m), 1.60 (1H, m), 1.32 (3H, d, J 6.5 Hz), 0.93 (9H, s), 0.89 (9H, s), 0.09 (3H, s), 0.07 (3H, s), 0.06 (3H, s), 0.04 (3H, s); δC (75 MHz, CDCl3) 172.6, 133.7, 124.7, 78.4, 74.5, 70.5, 34.6, 32.1, 25.8, 25.6, 22.7, 20.2, 18.3, 18.0, −4.8, −4.9, −5.1, −5.4; m/z (EI) 428.2777 (C22H44O4Si2 requires 428.2778, M+˙, 18%), 413 (28), 371 (76), 239 (81), 147 (69), 73 (100).
Compound 22
A magnetically stirred solution of methyl ester 20 (28 mg, 0.057 mmol) in EtOH (3 mL) was treated with NaOH (1 mL of a 2.5 M aqueous solution). The ensuing mixture was allowed to stir at 18 °C for 18 h, acidified to pH 3 (with 1 M aqueous HCl) then extracted with EtOAc (5 × 12 mL). The combined organic phases were washed with brine (1 × 5 mL) then dried (MgSO4), filtered and concentrated under reduced pressure to give acid 21 (26 mg, 95%) as a light-yellow oil. The crude acid was dissolved in THF (1 mL) containing Et3N (10 μL, 0.07 mmol). The resulting and magnetically stirred solution was treated with 2,4,6-trichlorobenzoyl chloride (9 μL, 0.06 mmol) and, after 2 h, diluted with toluene (70 mL). The ensuing mixture was then added, over ca. 10 minutes and via cannula, to a solution of DMAP (37 mg, 0.3 mmol) in refluxing toluene (10 mL). The resulting mixture was heated at reflux for 1 h then cooled, quenched with NaHCO3 (5 mL of a saturated aqueous solution) and extracted with EtOAc (3 × 20 mL). The combined organic fractions were washed with brine (1 × 5 mL) then dried (MgSO4), filtered and concentrated under reduced pressure. The light-yellow oil thus obtained was subject to flash chromatography (silica gel, 95 ∶ 5 v/v hexane–Et2O elution) and concentration of the appropriate fractions (Rf 0.3) then gave macrolactone 22 (22.5 mg, 89% from 20) as a clear colourless oil, [α]D −23 (c 0.2). νmax (KBr) 2932, 2858, 1723, 1649, 1466, 1364, 1254, 1159, 1074, 1003, 835, 777 cm−1; δH (300 MHz, CDCl3) 6.76 (1H, dd, J 15.8 and 4.7 Hz), 6.15 (1H, dd, J 15.8 and 1.6 Hz), 5.08 (1H, m), 4.43 (1H, m), 3.53 (1H, dm, J 8.9 Hz), 1.90–1.05 (10H, complex m), 1.28 (3H, d, J 6.6 Hz), 0.92 (9H, s), 0.90 (9H, s), 0.09 (3H, s), 0.06 (3H, s), 0.05(2) (3H, s), 0.04(8) (3H, s); δC (75 MHz, CDCl3) 168.4, 147.8, 121.7, 76.6, 75.7, 72.9, 32.2, 30.7, 27.7, 26.1, 26.0, 25.8, 23.4, 19.4, 18.5, 18.4, −4.4 (two signals superimposed), −4.6(7), −4.6(9); m/z (EI) 456.3090 (C24H48O4Si2 requires 456.3091, M+˙, 3%), 399 (28), 371 (13), 241 (34), 216 (51), 198 (62), 147 (88), 73 (100).Acknowledgements
We thank the Institute of Advanced Studies (IAS) and the Australian Research Council for financial support. FV is the grateful recipient of a 1999–2000 Summer Research Scholarship funded by the IAS.References
- (a) A. Hirota, A. Isogai and H. Sakai, Agric. Biol. Chem., 1981, 45, 799 Search PubMed; (b) A. Hirota, H. Sakai, A. Isogai, Y. Kitano, T. Ashida, H. Hirota and T. Takahashi, Agric. Biol. Chem., 1985, 49, 903 Search PubMed; (c) H. Hirota, A. Hirota, H. Sakai, A. Isogai and T. Takahashi, Bull. Chem. Soc. Jpn., 1985, 58, 2147 CAS.
- A. Hirota, H. Sakai and A. Isogai, Agric. Biol. Chem., 1985, 49, 731 Search PubMed (+)-Cladospolide B and (−)-isocladospolide B have also recently been isolated from the sponge-derived fungus Cladosporium herbarum ( C. J. Smith, D. Abbanat, V. S. Bernan, W. M. Maiese, M. Greenstein, J. Jompa, A. Tahir and C. M. Ireland, J. Nat. Prod., 2000, 63, 142 Search PubMed ). The full stereochemistry of (−)-isocladospolide B has recently been established by total synthesis: X. Franck, M. E. V. Araujo, J.-C. Jullian, R. Hocquemiller and B. Figadere, Tetrahedron Lett., 2001, 42, 2801 CrossRef CAS.
- Y. Fujii, A. Fukuda, T. Hamasaki, I. Ichimoto and H. Nakajima, Phytochemistry, 1995, 40, 1443 CrossRef CAS.
- H. Zhang, H. Tomoda, N. Tabata, H. Miura, M. Namikoshi, Y. Yamaguchi, R. Masuma and S. Omura, J. Antibiot., 2001, 54, 635 Search PubMed.
- (a) K. Mori and S. Maemoto, Liebigs Ann. Chem., 1987, 863 Search PubMed; (b) S. Maemoto and K. Mori, Chem. Lett., 1987, 109 CAS.
- I. Ichimoto, M. Sato, M. Kirihata and H. Ueda, Chem. Express, 1987, 2, 495 ( Chem. Abstr. , 1988 , 108 , 167167 ) Search PubMed.
- (a) G. Solladié, A. Almario and D. Carmen, Pure Appl. Chem., 1994, 66, 2159 CAS; (b) G. Solladié and A. Almario, Tetrahedron: Asymmetry, 1995, 6, 559 CrossRef CAS.
- For useful points-of-entry into the current literature on RCM processes leading to medium-sized rings see: (a) M. E. Maier, Angew. Chem., Int. Ed., 2000, 39, 2073 CrossRef CAS; (b) A. Fürstner, Angew. Chem., Int. Ed., 2000, 39, 3012 CrossRef CAS.
- M. G. Banwell and K. J. McRae, Org. Lett., 2000, 2, 3583 CrossRef CAS.
- For a comprehensive discussion on the production and synthetic utility of cis-1,2-dihydrocatechols, see: T. Hudlicky, D. Gonzalez and D. T. Gibson, Aldrichimica Acta, 1999, 32, 35 Search PubMed.
- (a) S. Hatakeyama, K. Satoh, K. Sakurai and S. Takano, Tetrahedron Lett., 1987, 28, 2713 CrossRef CAS; (b) S. Hatakeyama, K. Satoh, K. Sakurai and S. Takano, Tetrahedron Lett., 1987, 28, 2717 CrossRef CAS.
- M. G. Banwell, G. S. Forman and D. C. R. Hockless, Indian J. Chem., 1999, 38B, 1011 Search PubMed.
- (a) D. R. Boyd, N. D. Sharma, H. Dalton and D. A. Clarke, Chem. Commun., 1996, 45 RSC; (b) D. Gonzalez, V. Schapiro, G. Seoane, T. Hudlicky and K. Abboud, J. Org. Chem., 1997, 62, 1194 CrossRef CAS.
- M. V. R. Reddy, A. J. Yucel and P. V. Ramachandran, J. Org. Chem., 2001, 66, 2512 CrossRef CAS.
- (a) C. R. Johnson and H. Sakaguchi, Synlett, 1992, 813 CrossRef CAS; (b) C. Orrenius, N. Oehrner, D. Rotticci, A. Mattson, K. Hult and T. Norin, Tetrahedron: Asymmetry, 1995, 6, 1217 CrossRef CAS.
- E. L. Dias, S. T. Nguyen and R. H. Grubbs, J. Am. Chem. Soc., 1997, 119, 3887 CrossRef CAS.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS.
- M. Hikota, Y. Sakurai, K. Horita and O. Yonemitsu, Tetrahedron Lett., 1990, 31, 6367 CrossRef CAS Also see: I. Paterson, D.Y.-K. Chen, J. L. Aceña and A. S. Franklin, Org. Lett., 2000, 2, 1513 Search PubMed.
- For other examples of the synthesis of ten-membered rings by RCM, see: A. Fürstner and K. Radkowski, Chem. Commun., 2001, 671 Search PubMed and references cited therein.
- J. Inanaga, T. Katsuki, S. Takimoto, S. Ouchida, K. Inoue, A. Nakano, N. Okukuda and M. Yamaguchi, Chem. Lett., 1979, 1021 CAS.
- B. C. Ranu, U. Jana and A. Majee, Tetrahedron Lett., 1999, 40, 1985 CrossRef CAS.
- H. D. Maynard and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 4137 CrossRef CAS.
Footnotes |
| † All new and stable compounds had spectroscopic data (IR, NMR, mass spectrum) consistent with the assigned structure. Satisfactory combustion and/or high-resolution mass spectral analytical data were obtained for new compounds and/or suitable derivatives. |
| ‡ All optical rotations were determined in chloroform solution at 20 °C. |
| § Enantiomeric excesses for alcohols (+)-9 and (−)-9 were determined by GLC analyses of the derived acetates using a 25QC2/CYDEX-B 0.25 capillary column supplied by SGE International Pty Ltd (e-mail: info@sge.com.au). |
| This journal is © The Royal Society of Chemistry 2002 |