Stoichiometric asymmetric processes
Simon Jones
Department of Chemistry, University of Newcastle upon Tyne, Bedson Building, Newcastle upon Tyne, UK NE1 7RU
First published on 6th February 2001
Abstract
Covering: January 2000 to December 2000. Previous review: J. Chem. Soc., Perkin Trans. 1, 2001, 95.
1. Introduction
When reading and abstracting this ‘millennium’ edition of Stoichiometric Asymmetric Processes it was immediately apparent that oxazolidinone and chiral hydrazone auxiliaries are the most widely used in asymmetric synthesis and in many places their use has become so routine and commonplace that synthetic procedures are cited rather than given explicitly. However, aside from chiral borane reagents, there are significantly fewer chiral reagents used routinely, which may perhaps be attributed to the cost of such reagents. As advances in the preparation and uses of polymer supported reagents are made this may well change in the near future.The topic Stoichiometric Asymmetric Processes covers a huge range of chemical transformations when one considers that all diastereoselective reactions of non-racemic substrates containing a stereogenic centre fall under this heading, including areas as diverse as rearrangements and glycosylation reactions. Thus, following the lead of Peter O'Brien's contribution last year, I have decided to concentrate on those transformations whereby the stereodirecting group is or has the potential to be removed at the end of the synthetic scheme. I have surveyed a majority of the primary chemical literature from 2000 but this review is far from comprehensive. I have decided to keep to a similar format to that introduced last year and have concentrated on the applications that auxiliaries have been used for in organic synthesis rather than the fine details regarding their reactivity and selectivity. However, some things have changed, and I have removed the section on the use of chiral reagents and auxiliaries in synthesis and incorporated this into the sections relating to transformations of that auxiliary. I have also added a few more subsections to help categorise the transformations.
2. Chiral auxiliaries
2.1 α-Alkylation
Asymmetric alkylation of lithium and sodium enolates of oxazolidinone and camphor sultam auxiliaries is one the most widely used transformations for the preparation of synthetic targets. Such reactions have been employed in many synthetic sequences including the synthesis of renin inhibitor BILA 2157 BS on a 0.6 kg scale,1 both enantiomers of enterlactone,2 isotopically 13C and D labeled L-leucine and other α-amino acids,3,4 β-amino acids through a subsequent Curtius rearrangement,5 pitiamide A,6 (−)-eburnamonine and (+)-epi-eburnamonine,7 the sex pheromone of the sandfly Lutzomyia longipalpis,8 α,α′-cis and trans-disubstituted medium ring ethers,9 (R)-2-propyloctanoic acid,10 orthogonally protected tricarballylic acid esters,11 the terminal acid portion of zooxanthellatoxins,12 epothilone B,13 the C1–C25 domain of sanglifehrin A,14 β2-amino acids15 and (1S,3S,7R)-3-methyl-α-himachalene, the sex pheromone of the sandfly Lutzomyia longipalpis.16 A few other interesting examples of oxazolidinone enolate alkylation stand out; alkylation of the lithium enolate of an oxazolidinone with a triflate electrophile gave a 3 ∶ 1 mixture of C-alkyl and O-alkyl products, the former as a single diastereomer.17 This was reported as the first example of O-alkylation in oxazolidinone enolate alkylation. Oxidative self coupling of lithium enolates of N-acyloxazolidin-2-ones with reagents such as TiCl4 have been shown to occur with good yields and levels of selectivity.18 Sibi and Rheault have investigated the stereoselectivity of the radical allylation of α-bromo-N-acyloxazolidinones 1 with allyltin reagents (Scheme 1).19 Quite surprisingly, higher selectivities were obtained at higher temperatures and a working model was proposed based upon the rotational barriers of the chelated and non-chelated radical intermediates. | ||
| Scheme 1 | ||
Although extensively used in synthesis, work is still carried out aimed at improving the efficiency and/or selectivity of oxazolidinone auxiliaries. Perhaps the most well known examples of these are the SuperQuat auxiliaries developed by Davies. Bull et al. have demonstrated that enhanced stereoselectivity in the alkylation of lithium enolates of the valine-derived SuperQuat chiral auxiliary is due to the geminal methyl groups controlling the conformation of the isopropyl stereodirecting group.20 This in effect makes the isopropyl group behave like a tert-butyl group as far as controlling the diastereoselectivity of the alkylation step. Asymmetric alkylation of benzyl SuperQuat auxiliaries has been demonstrated for a number of substrates and electrophiles and the products were subsequently cleaved to the α-substituted aldehydes using DIBAL-H with no loss of stereochemical integrity.21 The first C2 symmetric bis(oxazolidinone) auxiliary 2 has been prepared and undergoes asymmetric alkylations using methyl iodide with facial selectivities at each enolate of 95 ∶ 5 (Scheme 2).22 The auxiliary also has excellent water solubility making recycling through aqueous extraction a viable option.
 | ||
| Scheme 2 | ||
Other analogues of oxazolidinones have also been used but to a lesser extent. The diastereoselectivity of the alkylation of 2-imidazolidinone derivatives was found to be dependent upon the second nitrogen substituent,23 while alkylation of the enolate of a glycinamide imidazolidinone 3 has been used in the diastereoselective synthesis of α-amino acids (Scheme 3).24
 | ||
| Scheme 3 | ||
Asymmetric alkylation of a dienolate provides quick access to chiral α-substituted β-alkenes, although this methodology has not been used to as great an extent as enolate alkylation. Application of this methodology has been demonstrated in the synthesis of 1α-hydroxyvitamin D525 and in the synthesis of a fragment of madindolines26 using oxazolidinones as the stereodirecting group.
Another preferred method for preparation of chiral α-substituted carbonyls is through the alkylation of aza-enolates of chiral hydrazone auxiliaries, which has been applied to the synthesis of (+)-maritimol,27 stigmolone,28 indolizidine alkaloids,29 and components from crocodile exocrine secretion.30 Ring opening reactions of p-tolylsulfonylaziridines with aza-enolates derived from the hydrazone 4 proceeded in excellent diastereoselectivity and have been used in the synthesis of γ-amino nitriles 5 and ketones 6 (Scheme 4).31,32
 | ||
| Scheme 4 | ||
The use of axially chiral reagents and catalysts continues to expand. Fujita et al. have reported in depth studies of the reaction of enolates derived from axially chiral pyrrolidinones 7 with a series of electrophiles (Scheme 5).33 The pyrrolidinones were prepared in excellent yield and with apparent complete chirality transfer and underwent reaction with electrophiles with excellent selectivity.
 | ||
| Scheme 5 | ||
Clayden et al. have demonstrated that amides possessing axial chirality may be deprotonated and quenched with a variety of electrophiles giving good yields and reasonable selectivities (69% ee).34 These compounds could be converted to chiral atropisomers bearing no stereogenic centers in a short synthetic sequence.
Terpenes have been used as a framework for chiral auxiliaries. A novel pinene-based auxiliary has been prepared and applied to asymmetric alkylations with poor to moderate diastereoselectivities,35 while the diastereoselective ring opening of a chiral epoxide with the enolate of a hydroxypinanone auxiliary has been investigated using a variety of bases and additives.36 Diastereoselective alkylation of a terpene-derived aza-enolate has been shown to proceed in excellent stereoselectivity and has been applied to the synthesis of 4,5-dihydroxypipecolinic acid.37 A conformationally restricted metabotropic glutamate receptor agonist bearing a cyclopropyl group has been prepared by dialkylation of a bis(menthyl ester)38 and asymmetric cyclopropanation has also been carried out by a tandem alkylation–Darzens reaction of a pyridinium salt 8 bearing the 8-phenylmenthyl auxiliary (Scheme 6).39
 | ||
| Scheme 6 | ||
The preparation of amino acid derivatives still continues to attract attention. One of the more widely used methods has been alkylation of Schöllkopf's bislactim ether template that has been used in the synthesis of 3-heteroaromatic substituted alanines,40 tryptophan analogues,41 silaproline,42 (+)-deoxypyridinoline,43 and α-methyl-α-substituted amino acids using mild phase transfer conditions.44 Chiral nickel(II) complexes of Schiff bases 9 have been used for the preparation of amino acids by alkylation of alanine and glycine templates (Scheme 7), which was amenable to large scale synthesis.45 This methodology was applied to the synthesis of ω-borono-α-amino acids as active site probes of arginine NO synthase46 and in the synthesis of 2′,6′-dimethyltyrosine via complex 10.47
 | ||
| Scheme 7 | ||
Stereoselective alkylation of aldimines using chiral ansa pyridoxyl derivatives proceeded in good enantiomeric excess, while introduction of a second stereogenic element allowed double asymmetric induction and greatly increased selectivities.48 Other methods to prepare amino acids include the alkylation of Williams' glycine template to prepare (2R,5R)- and (2S,5R)-5-hydroxylysine,49 alkylation of a chiral morpholinecarboxylate to prepare a non-proteinogenic fluorescent amino acid,50 alkylation of a novel chiral oxazoline with a range of active electrophiles,51 alkylation of a chiral aza-enolate with 1,2-fluorobromides,52 and an alkylation–cyclisation procedure to prepare α,α′-diaminodicarboxylic acids.53,54
Many other chiral auxiliaries have been investigated in alkylation reactions. Some in particular merit further details. Denmark and Kim have reported good to excellent diastereoselectivities in the alkylation of phosphonamide auxiliaries based on a C2 symmetric diamine.55 Hanessian and Cantin have demonstrated that γ-chlorodienolates of similar chiral phosphonamides undergo a highly diastereoselective Darzens-like reaction with glyoxylaldehyde-derived oximes.56 Seebach's self-replication of chirality is a useful procedure that has been used to prepare substrates for RCM reactions,57 in the synthesis of both enantiomers of a neuroexcitant58 and both enantiomers of the pheromone of the tropical wandering spider Cupiennius salei11 (Scheme 8).59
 | ||
| Scheme 8 | ||
Diastereoselective alkylation of chiral oxazolidines has been used in the synthesis of the ladybird defence alkaloid (+)-calvine and (+)-2-epicalvine,60 a compound from the pheromone gland of the stinkbug,61 and 3-alkyl-3-arylpyrrolidines.62 Alkylation of sodium enolates of sulfonamides with α-bromocarboxylates appeared to give single diastereomers of addition products that were subsequently reduced to diols.63 This is interesting since the bromocarboxylates used were racemic implying that racemisation of such reagents is fast under these reaction conditions.
The chemistry of arene chromium tricarbonyls has been further expanded and the first example of the addition of the enolate of tert-butyl acetate to a chromium arene tricarbonyl complex to give a cyclohexadiene product has been reported with excellent levels stereoselectivity.64 Huang and Comins achieved the highly diastereoselective addition (de >95%) of a zinc enolate equivalent to a trans-2-(α-cumyl)cyclohexanol pyridinium derivative, the key step in the synthesis of (+)-streptazolin.65 Chiral alanine dianions have been shown to give excellent levels of selectivity in some alkylation reactions66 and when employed with ethyl bromoacetate, a built-in ‘auxiliary release’ was used to free the chiral auxiliary. Chiral aldehydes have been used to prepare non-racemic aminals with good diastereoselection (84 ∶ 16)67 and N-acyl derivatives of these have subsequently been used in enolate alkylations giving diastereoselectivities of over 90%. The diastereoselectivity of the allylation of enolates of bicyclic oxazolidines was found to be dependent upon the nature of the protecting group of the amino alcohol.68
Other auxiliaries that have been used in asymmetric alkylations include a thiazolo[2,3-a]isoindolin-1-one,69 α-methylbenzylamine70 and phenylglycinol71 in the synthesis of tetrahydroisoquinolines, an 8-phenylmenthyl ester in an intramolecular radical cyclisation,72 a BINOL ester in the synthesis of (+)- and (−)-ferruginol,73 2-phenylcyclohexanol in the synthesis of (+)-huperzine A,74N-methylephedrine in the synthesis of the C14–C25 portion of amphidinolide B1,75 and O-methyl phenylglycinol in the deprotonation–alkylation of oxazolidines76 and aziridines.77
2.2 Aldol reactions and related processes
As with asymmetric alkylation, highly diastereoselective syn-aldol condensations using chiral boron enolates of oxazolidinones generated using dibutylboron triflate and an amine base are now used as a matter of routine in synthetic schemes. Although a robust model for the selectivity observed in this reaction is well established, the first report of the structure of a stable boron enolate of an oxazolidinone has been reported using 600 MHz 2D NOESY NMR and molecular modeling techniques.78 Since the substitution pattern of the aldol product obtained closely matches that of many natural products, application of this methodology to total syntheses has been extensive. Successful uses include the synthesis of (+)-rajadone,79 the C29–C51,80 C29–C4581 and C29–C4482 fragments of spongistatin 1, the dihydrobenzofuran segment of ephedradine C,83 a pyrrolidine α-glycosidase inhibitor,84 antillatoxin,85 the C1–C9 fragment of rhizoxin,86 hapalosin,87 (+)-madindoline A and (−)-mandinoline B,88 (−)-CP-263,114,89 and (+)-discodermolide.90 Boron enolates of oxazolidinones have also been used to prepare substrates for stereoselective phenylsulfanyl rearrangements to provide spiro[4.5]decanes.91 Reactions of boron dienolates are not as widespread as those of enolates. A boron dienolate of an oxazolidinone 12 has been shown to react with a chiral aldehyde 13 to give the desired aldol product 14 as a 1 ∶ 1 mixture of Z and E isomers that was used in the synthesis of the tris(oxazole) macrolide ulapualide A92 (Scheme 9) and similarly in the synthesis of polyene macrolides.93 | ||
| Scheme 9 | ||
One of the advantages of aldol reactions of oxazolidinones is that syn- and anti-aldol products may be selected by changing the reaction conditions and examples of this have been used in the synthesis of ribavirin94 and phoboxazole B.95
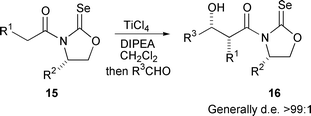
Although highly successful, aldol reactions are obviously not limited to boron enolates of oxazolidinones. Titanium(IV) enolates of N-acyloxazolidine-2-selones 15 give predominately ‘non-Evans’ syn-aldol products 16 with excellent diastereoselectivity,96 and the selone was found to play a pivotal role in determining the stereoselectivity (Scheme 10).
 | ||
| Scheme 10 | ||
Titanium enolates have also been used with oxazolidinones and oxazolidinethiones that undergo syn-selective aldol reactions with the best selectivities observed for the oxazolidinethione97 which was applied to the synthesis of (+)-prelaureatin and (+)-laurallene. Such enolates have also been shown to react in the presence of (−)-sparteine to give excellent diastereoselectivity in favour of the syn-aldol product and used in the synthesis of carbocyclic nucleosides.98 Tin enolates have been used to mediate an aldol reaction of an α-hydroxyacyloxazolidinone giving the anti-aldol product in good diastereoselectivity when treated with a chiral aldehyde.99
Use of acetyl enolates in asymmetric aldol reactions is notoriously problematic and various methods have been employed to overcome this. A study of the use of a number of boron triflates and α-thioacetyl derivatives attempted to address this problem and was applied to the C1–C6 segment of epothilones.100 Other types of enolate other than boron have also been investigated. An acetyl tin enolate of a thiazolidine-2-thione has been shown to give aldol products with excellent diastereomeric excess in the synthesis of (−)-teubrevin G,101 while intermolecular SmI2-promoted Reformatsky-type reactions of a number of acetyloxazolidin-2-one derivatives 17 has been investigated with good levels of diastereoselectivity (Scheme 11).102
 | ||
| Scheme 11 | ||
Examples of transformations of new oxazolidinone auxiliaries continue to be developed. The asymmetric boron-mediated aldol reactions of a new glucose-derived oxazolidinone 18 have been described giving diastereomeric ratios from 3 ∶ 1 to 16 ∶ 1 in favor of the expected syn-product 19 depending upon the protecting groups of the carbohydrate and the aldehyde employed (Scheme 12).103
 | ||
| Scheme 12 | ||
Asymmetric aldol reactions are not limited to employing oxazolidinones or thio-substituted analogues as chiral auxiliaries and many other examples exist that make use of the formation of an amide-type bond in the addition and cleavage steps. Boron enolates of imidazolidin-2-ones104 and imidazolidinone glycine equivalents105 undergo highly diastereoselective syn-selective aldol reactions with good to excellent diastereoselectivities. Sultams are often comparable in their chemistry to oxazolidinones and have been used in boron-mediated aldol reactions with chiral aldehydes and α-ketoesters.106 Although the former proceeded with total diastereoselectivity in favour of the syn-product, the ketoester gave a 3 ∶ 1 mixture to two diastereomers. Titanium enolates of sultams have also been employed giving the anti-aldol product in excellent yield (95%).107 Amides derived from pseudoephedrine 20 have been used in asymmetric aldol reactions in the presence of different metal cations, in particular zirconocene dichloride gave selectivities of over 99 ∶ 1 in favour of the syn-isomer 21 (Scheme 13),108 and this auxiliary was used in the first asymmetric synthesis of isoflavanones.109 The aldol reactions of N-propionylprolinol derivatives have also been investigated with and without the addition of Lewis acids.110
 | ||
| Scheme 13 | ||
A number of auxiliaries make use of the formation of esters and acetals to facilitate the attachment or cleavage steps. Paterson et al. have made use of the excellent diastereoselective control in the aldol reaction of boron enolates derived from chiral ketones in an auxiliary type fashion.111 Thus, after the desired aldol reaction had taken place, oxidative cleavage of the stereodirecting group was achieved, although this destroyed the stereochemistry of the attached chiral auxiliary.
Relatively few examples of aldol reactions with terpene-derived auxiliaries have been reported. Thus, menthol- and camphor-derived acetylenic esters 22 have been used in titanium-mediated additions giving Baylis–Hillman products 23 with good regioselectivities and varying diastereoselectivities (Scheme 14).112 The best auxiliary was found to be the alcohol derived from D-camphor giving diastereoselectivities >97 ∶ 3 for a range of structurally diverse aldehydes.
 | ||
| Scheme 14 | ||
Camphor derivatives have also been used as the stereodirecting group in a Darzens reaction of α-bromoacetyl substrates giving epoxides of >97 ∶ 3 diastereoselectivity,113 while a novel pinene-based auxiliary has been prepared and used in asymmetric aldol reactions with poor to moderate diastereoselectivities.35 Aldol reactions of aza-enolates derived from the hydroxypinanone derivative 24 have been carried out using a catalytic quantity of base giving good diastereocontrol at the α-carbon but essentially no control at the β-centre (Scheme 15).114
 | ||
| Scheme 15 | ||
Seebach's self-reproduction of chirality has been demonstrated on a 100 g scale in the synthesis of (S)-oxybutynin115 and in the synthesis of a chiral spiro aminochromane,116 the latter giving poor levels of stereocontrol at the newly formed stereogenic centre.
Boron and titanium enolates of chiral α-chloromethyleneoxazolines give oxazolinyl oxiranes when treated with ketones in excellent diastereoselectivity.117 Other asymmetric aldol processes include the reaction of dienolates derived from chiral oxazolidines with aldehydes,118 a diastereoselective Mukiyama aldol reaction of a chiral furyl sulfoxide,119 the synthesis of enantiomerically pure mono- and bis-epoxides of isoprene using Ley's dispoke protected lactate,120 and addition of (E)-enolates to chiral dithiane ketone equivalents.121
2.3 Michael additions
Michael additions are particularly important transformations as they facilitate bond formation β to a carbonyl centre and often allow construction of quaternary carbon centres. Examples of this include the use of a chiral aza-enolate in the first steps of the synthesis of some polycyclic diterpenes122 and chiral enamines 25 derived from β-oxoesters that react under copper catalysis at ambient temperature and without an inert atmosphere (Scheme 16).123 | ||
| Scheme 16 | ||
Addition of cuprate and organocopper reagents to chiral acceptors is a well used method for performing conjugate additions. Lithium alkyl cyanocuprates undergo highly diastereoselective conjugate addition from the least hindered exo-face to chiral bicyclic lactams and this has been applied to the synthesis of trans-3,4-disubstituted piperidines.124 The addition of various cuprate and organocopper reagents to unsaturated tert-leucine oxazolidinone acceptors has been investigated with and without an added Lewis acid with excellent levels of diastereoselectivity.125 Similar acceptors have been used in the synthesis of kalkitoxin,126 an α,β-substituted histidine analogue127 and 3′O,4′O-dimethylfuniculosin (Scheme 17).128
 | ||
| Scheme 17 | ||
Organocopper and organoaluminium reagents have also been used in domino Michael–aldol and Michael–Mannich reactions with chiral oxazolidinones to construct highly functionalised cyclohexane frameworks.129 In an interesting switch, organolithium reagents based on chiral oxazolidinones 26 undergo copper-mediated Michael addition to an unsaturated aldehyde with excellent levels of diastereocontrol (Scheme 18).130
 | ||
| Scheme 18 | ||
Addition of organocopper reagents to unsaturated chiral phosphonate derivatives gave good to poor levels of diastereoselectivity.131 Under appropriate circumstances, organolithium and magnesium reagents can also add to chiral acceptors without the aid of copper additives. Diastereoselective Michael addition of MeLi to a vinyl selenide bearing a cyclohexanol auxiliary followed by quenching with an alkyl halide gave >15 ∶ 1 diastereoselectivity.132 Highly diastereoselective Grignard addition–alkylation to chiral aryloxazolines and imines have been shown to occur in good yield via an unusual Michael addition of the nucleophile directly to an aromatic ring.133
Carbon-based nucleophiles derived from enolates or enolate equivalents are generally more useful in Michael additions than alkyllithium and magnesium reagents since the conditions for their generation are invariably less harsh. Enolates themselves have been used in the synthesis of a key intermediate of a muscarinic M-3 receptor antagonist,134 the novel H3 agonist Sch 50971135 and in the synthesis of pyroglutamates.136 An interesting example of an asymmetric intramolecular tandem Michael–aldol reaction of an enolate with phenylmenthyl enoates 27 gave tricyclic structures 28 with excellent levels of diastereoselectivity (Scheme 19).137
 | ||
| Scheme 19 | ||
Glycine-derived enolates have been used in the synthesis of pyroglutamates,138 and nickel(II) Schiff base complexes of such enolates found to react with α,β-unsaturated oxazolidin-2-one acceptors in excellent yield and diastereoselectivity.139 The origin of the selectivity of this reaction was investigated in some detail and the stereochemical outcome was found to be independent of the stereochemistry of the Schiff base.140 Aza-enolates generated from chiral imines have been shown to undergo Michael additions with excellent levels of diastereoselectivity141 and used in the synthesis of (+)-α-vetivone.142 α-Substituted δ-lactams 30 have been prepared via Michael addition of the enolate of a chiral hydrazone 29 to nitro alkenes with good to excellent levels of stereocontrol (Scheme 20).143
 | ||
| Scheme 20 | ||
A study of the conjugate addition of an aza-enolate of Schöllkopf's bislactim ether to (E)- and (Z)-prop-1-enylphosphonates was found to occur with complete diastereoselectivity at the α-centre and excellent selectivity at the β-centre in all cases except when HMPA, SnCl2 or CuBr2 were used.144 In a related reaction, Michael addition of a nitro enolate to a chiral chromium tricarbonyl Michael acceptor proceeded with excellent diastereocontrol.145 Enolate equivalents are often prepared and reacted under milder conditions than enolates themselves and in this context enamines are very useful reagents as they can be prepared using chiral amines. A phenyloxazolopiperidine derivative 31 has been prepared and used as a latent enamine equivalent in an asymmetric Michael addition with methyl vinyl ketone giving the addition adduct 32 as a single diastereomer in reasonable yield (Scheme 21).146
 | ||
| Scheme 21 | ||
Enantioselective Michael additions of enamines to nitro olefins have been shown to occur with excellent yield and degrees of diastereoselectivity and the products from these reactions converted into non-racemic pyrrolidines.147
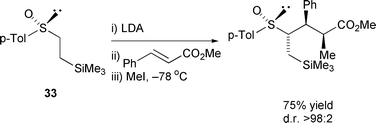
Other miscellaneous enolate equivalents have been used to perform carbon–carbon bond formation using Michael additions. 2,6-Dimethylmorpholine has been used as a chiral auxiliary in the Michael addition of Fischer carbene complexes to unsaturated nitro compounds in 75% de and 90% isolated yield.148 Highly stereoselective conjugate additions of α-carbanions of chiral sulfoxides 33 to unsaturated esters followed by quenching with alkyl halides and aldehydes has been reported (Scheme 22),149 while C2 symmetric sulfoxides have been shown to add to stabilised Michael acceptors giving a single diastereomer of addition product.150
 | ||
| Scheme 22 | ||
A diastereoselective Michael–Darzens-type addition of a sulfur ylide to an unsaturated chiral sultam has been used to prepare the pheromone of the pine sawfly.151 Hanessian et al. have demonstrated phosphonamide dianions undergo highly diastereoselective Michael additions with ketones, lactones and lactams,152 while phosphonamide anions have been used in a tandem Michael addition–Darzens-type type reaction to prepare cyclopropanes.153 The asymmetric Michael addition of alkyl radicals has not received as much attention as other carbon–carbon bond forming processes, although a few reports have been made of addition to unsaturated chiral sulfoxides,154 a crotonyl group tethered to a carbohydrate in the presence of diethylaluminium chloride155 and of photosensitized radical addition to chiral fumaric acid derivatives156 with good diastereoselectivities.
Michael additions are not limited to the formation of carbon–carbon bonds alone and additions of nitrogen nucleophiles are quite commonplace. One of the best-known examples of such reactions is the addition of homochiral ammonia equivalents developed by Davies. This work has been extended through the development of a new route towards chiral Baylis–Hillman products 35 by the diastereoselective Michael addition of a chiral lithium amide 34, followed by subsequent stereoselective boron-mediated aldol reaction and finally Cope elimination (Scheme 23).157
 | ||
| Scheme 23 | ||
Bull et al. have provided further examples of chiral lithium amide additions to Michael acceptors in the preparation of substrates to demonstrate the debenzylation reactions of amines.158 Other applications of this methodology have been reported including the synthesis of β-haloaryl β-amino acids,159 cyclic β-amino acids,160 β-homolysine,161 plakoridine A,162 trisubstituted piperidines163 and the dihydrobenzofuran segment of ephedradine C.83 Chiral hydrazines have also been used as ammonia equivalents and employed in the asymmetric synthesis of α-substituted β-amino sulfones164 and in a stepwise Michael addition–α-alkylation of unsaturated lactones.165 Michael addition of a chiral oxazolidinone 36 to a nitro olefin gave a good ratio of anti ∶ syn-addition products 37 (85 ∶ 15) that were used in the synthesis of dethiobiotin (Scheme 24).166
 | ||
| Scheme 24 | ||
Asymmetric Michael addition of amines has also been carried out using chiral acceptors. Addition of resin-bound amino acids to a chiral oxazolidinone acceptor proceeded in good to poor diastereoselectivity,167 while the synthesis of enantiomerically pure aziridines 39 was accomplished by a fairly diastereoselective (80 ∶ 20) addition of a hydroxylamine to a chiral imidazolidinone acceptor 38, followed by separation and cyclisation (Scheme 25).168
 | ||
| Scheme 25 | ||
Addition of amine and alcohol nucleophiles to chiral α,β-unsaturated sulfamines has been shown to occur with good levels of diastereoselectivity, although this is limited in scope as the nucleophile must be used as the solvent.169
Oxygen-based nucleophiles have been used less often in such reactions. Two consecutive diastereoselective Michael additions of a propargylic (prop-2-ynyl) alcohol to a chiral unsaturated sulfoxide have been used in the synthesis of (+)-asteriscanolide.170 Oxidative cyclisation of chiral phenolic oxazolines 40 in the presence of iodobenzene diacetate gave an intermediate alkoxyspirolactam 41 that underwent spontaneous intramolecular Michael addition to give a single diastereomer of a tricyclic compound 42 (Scheme 26).171
 | ||
| Scheme 26 | ||
As with oxygen nucleophiles, relatively few examples of sulfur nucleophiles in Michael additions have been reported. Of particular interest is a novel tandem Michael addition and Meerwein–Ponndorf–Verley reduction which has been carried out using the 10-mercaptoisoborneol auxiliary 43 giving alcohols 44 in high yields and excellent levels of stereoselectivity (Scheme 27).172,173
 | ||
| Scheme 27 | ||
The diastereoselectivity of the addition of thiols to camphor-derived Michael acceptors has been investigated giving a wide range of selectivities and yields depending upon the reaction conditions.174 Addition of a thiol to cyclic acceptors derived from 3-hydroxybutyric acid gave inseparable mixtures of diastereomers, the selectivity of which depended upon the exocyclic alkyl group.175
Two other unusual Michael additions warrant attention; Enders et al. have developed new methods for the synthesis of α-substituted β-nitrophosphonic acids using a chiral phosphite complex derived from 1,1,4,4-tetraphenyl-2,3-O-isopropylidene-L-threitol (TADDOL).176 Reaction of such diethylzinc-activated complexes with a series of nitro alkenes afforded the desired products in good yield and diastereomeric excess with cleavage of the TADDOL auxiliary giving the nitrophosphonic acids. Degnan and Meyers have reported the only example in this survey of the highly diastereoselective conjugate addition–alkylation of silyl anions to naphthyloxazolines 45 and their application to the synthesis of aminotetralin (Scheme 28).177
 | ||
| Scheme 28 | ||
2.4 Addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O and CN bonds
O and CN bonds
Stereocontrolled addition to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN bonds represents an easy route to the introduction of an additional stereogenic centre, and when using enolates or their equivalents as nucleophiles, the formation of a carbon–carbon bond. Oxazolidinone chiral auxiliaries have again been used extensively, primarily in the addition to CN bonds. Various substitution of N-acyl groups has been investigated in some detail to study the addition of enolates of oxazolidinones to iminium ions generated from piperidines.178 New methodology has also been reported for the synthesis of β-amino acids by the addition of enolates of N-acyloxazolidinones 46 to N-acyloxyiminium ions generated in situ (Scheme 29).179 The reactions proceeded with moderate levels of selectivity and the main advantage of this
methodology is that chiral pyrrolidine and piperidine derivatives are easily prepared.
O and CN bonds represents an easy route to the introduction of an additional stereogenic centre, and when using enolates or their equivalents as nucleophiles, the formation of a carbon–carbon bond. Oxazolidinone chiral auxiliaries have again been used extensively, primarily in the addition to CN bonds. Various substitution of N-acyl groups has been investigated in some detail to study the addition of enolates of oxazolidinones to iminium ions generated from piperidines.178 New methodology has also been reported for the synthesis of β-amino acids by the addition of enolates of N-acyloxazolidinones 46 to N-acyloxyiminium ions generated in situ (Scheme 29).179 The reactions proceeded with moderate levels of selectivity and the main advantage of this
methodology is that chiral pyrrolidine and piperidine derivatives are easily prepared. | ||
| Scheme 29 | ||
Other types of chiral auxiliary have also been used. Enantioselective reaction of lithium enolates of sultam chiral auxiliaries with diphenylphosphinyl imines gave excellent diastereo- and enantioselectivities.180 A camphor-derived auxiliary has been used to demonstrate the versatility of an asymmetric Mannich-type reaction giving good to excellent yields and diastereoselectivity.181 One report described the addition of an enolate to a chiral sulfinyl imine auxiliary in the synthesis of all four stereoisomers of 4-hydroxypipecolic acid.182
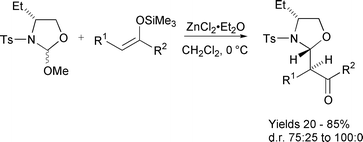
Enolate equivalents, in particular silyl enol ethers, have also been employed extensively. Addition of silyl enol ethers to chiral imines generated in situ gave good to excellent levels of diastereocontrol, the products of which could be transformed themselves into silyl enol ethers that underwent subsequent aldol reaction with excellent degrees of stereoselectivity.183 Further applications of this methodology have also been reported (Scheme 30).184
 | ||
| Scheme 30 | ||
Ytterbium triflate has been used to catalyse the addition of chiral silyl ketene aminals to imines and the diastereomeric ratio was found to be dependent upon the structure of the imine.185 A similar reaction has also been carried out under Mannich-type conditions.186 Glycosyl bromides have been used to activate Schiff bases to nucleophilic attack of silylketene acetals.187 Good yields were obtained but with moderate levels of diastereoselectivity (dr generally 3 ∶ 1) which was shown to be due to E to Z isomerisation of the CN bond after activation.
α-Carbanions of chiral sulfoxides have been used as chiral carbon nucleophiles. Thus, lithiation of the benzylic position of a chiral sulfoxide followed by quenching with a chloroformate, aldehyde or ketone gave the desired products in excellent diastereomeric excess.188 In the case of prochiral ketones and aldehydes, good to poor ratios of epimeric alcohols were formed. Such carbanions have also been shown to add in a matched/mismatched fashion to chiral N-sulfimines 47 (Scheme 31).189 In the matched case a diastereomeric excess of over 98% in 99% yield was observed.
 | ||
| Scheme 31 | ||
Although in general significantly more reactive, organometallic reagents undergo facile addition to CO and CN bonds, sometimes with the added benefit of chelation to improve stereochemical control. In particular, addition of Grignard reagents to chiral imines has been widely used, for example, in the synthesis of 4-hydroxypipecolic acid by addition to a chiral pyridinium salt,190 in the preparation of chiral substituted piperidines,191 and in the three step synthesis of an HIV non-nucleoside reverse transcriptase inhibitor.192 An in depth study has been carried out using allylmagnesium and zinc reagents with bisimines 48 bearing the α-methylbenzylamine stereodirecting group (Scheme 32)193 and independent studies have shown that dynamic kinetic resolution may
be observed with Grignard additions to such species.194
 | ||
| Scheme 32 | ||
Grignard addition to chiral aza-acetals proceeded with reasonable levels of stereoselectivity dependent upon the Grignard reagent,357 and reactions with related C2 symmetric bisoxazolidines have also been investigated.195 An in depth study of the enantioselectivity of the addition of Grignard reagents to chiral sulfinyl imines has also been carried out.196 Nakamura et al. have investigated the diastereoselective addition of Grignard reagents to axially chiral aryl sulfoxide aldehydes 49,197,198 the selectivity of which was dependent upon the structure of the sulfoxide and any additive (Scheme 33). Attempted addition of silyl enol ethers gave poorer diastereoselectivities.
 | ||
| Scheme 33 | ||
The only surveyed report of the asymmetric addition of a Grignard reagent to a ketone has been the addition of 2-naphthylmagnesium bromide to a menthyl pyruvate ester with poor levels of diastereocontrol.199
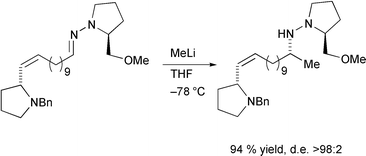
Organolithium reagents have also been employed with good degrees of success. Studies of the addition of such reagents to chiral 1-ferrocenylalkanimines,200 bisimines bearing the α-methylbenzylamine stereodirecting group,201 chiral oxazolidines,202 and aldehydes with axial chirality203 have all been reported. Addition of organolithium reagents to chiral hydrazones provides excellent degrees of stereocontrol and has been used in the synthesis of a defence alkaloid of the Mexican bean beetle (Scheme 34)204 and (+)-2-epideoxoprosopinine.205
 | ||
| Scheme 34 | ||
The application of heterosubstituted organolithium species has led to some success, for example, in the addition of lithium carbanions of chiral thiazolidines to aldehydes206 and lithiated allyl sulfones to chiral N-sulfinylamines.207
Other organometallic species, primarily organozinc and titanium reagents have been used, presumably due to their availability. Alexakis et al. have prepared a new C2 symmetric diamine in excellent enantiomeric excess by using a diastereoselective addition of allylzinc bromide to an α-methylbenzylamine-derived imine.208 Reaction of chiral imines with alkenyltitanium209 and allyltitanium alkoxides210 have been shown to proceed with good levels of diastereoselectivity. Addition of an allylindium reagent generated in situ to an activated ketone bearing a chiral amine gave excellent levels of diastereoselectivity.211
Reduction of CO and CN bonds provides one of the easiest methods to install a new stereogenic centre. Reduction of chiral imines with zinc borohydride has been extensively studied,212 and other borane- and alane-derived reducing agents have been used in the synthesis of chiral ferrocenyl amines,213 1,2-amino alcohols,214 tetrahydroisoquinolines,71 and piperidines.215 Related hydrazones have been reduced with LiAlH4 with varying levels of stereocontrol using 2-aminobutan-1-ol as an auxiliary.216
Chiral keto sulfoxides have been used widely as substrates for asymmetric reductions. The reduction of such γ-keto sulfoxides with and without addition of Lewis acids has been investigated for a number of substrates giving excellent yields and levels of diastereoselectivity.217 Reduction of a β-keto sulfoxide with DIBAL-H gave >95% diastereomeric excess that could be completely inverted by addition of ZnI2 to the reaction.218 Such reductions have been used in the synthesis of chiral 2-amino alcohols219 and in the synthesis of colletol (Scheme 35).220
 | ||
| Scheme 35 | ||
Fewer reports exist for the stereoselective reduction of ketones, presumably as there are now very efficient catalytic methods available for this transformation. The conditions for the stereocontrolled reduction of a chiral aziridinyl ketone have been optimised and used in the synthesis of threo-β-hydroxy-L-glutamic acid221 and in the preparation of both enantiomers of 4-hydroxycyclohex-2-enone, an important chiral building block, achieved by using a 1,2-reduction of the chiral cyclohexenone acetal.222
Some reports have been made on diastereoselective radical or radical-type addition to chiral carbonyls and imines. N-Acylhydrazones based on oxazolidinones have been prepared and have been shown to undergo highly diastereoselective radical addition to give N-acylhydrazines223 and sultam auxiliaries have been used to control the stereoselectivity of the carbon radical addition to oxime derivatives 50 (Scheme 36).224 These reactions generally proceeded in good yield and selectivity (86 ∶ 14 to 96 ∶ 4) and the products 51 were easily converted to α-amino acids.
 | ||
| Scheme 36 | ||
Triethylborane and diethylzinc-mediated radical additions to chiral glyoxylate and cyclic imines gave good selectivities with cyclic compounds and poorer selectivities with open chain substrates.225 2-Methoxymethylindolinone has been used as a chiral auxiliary in SmI2-mediated pinacol couplings to give substituted tartrate derivatives of high enantiomeric purity,226 while 8-phenylmenthol has been used as an auxiliary in normal and crossed pinacol couplings with reasonable levels of diastereoselectivity.227
The asymmetric Strecker reaction is a useful method to prepare chiral amino acids and several methods have been reported that employ this methodology, including the synthesis of the Corey precursor of lactacystin.228 Use of chiral sulfimines as substrates for this reaction has been disclosed229,230 and employed in the synthesis of diaminopimelic acids.231 A number of carbohydrate derivatives have been examined in asymmetric Strecker reactions using α-methylbenzylamine as the stereodirecting group.232 Diastereomeric ratios from 1 ∶ 1 to 7 ∶ 1 were obtained depending upon the substrate. An asymmetric Strecker reaction has been investigated in some detail with a series of chiral imines based on a racemic cyclopentane framework with varied levels of diastereoselection.233
Other miscellaneous additions to CO and CN bonds include the use of 1-phenylethylamine in the discrimination of enantiotopic symmetric bisketenes,234 stereoselective acetal formation in the desymmetrisation of glycerol using camphor sulfamides235 and in the synthesis of muricatetrocin C,236 the stereoselective synthesis of trans-aryl vinyl epoxides using a chiral sulfur ylide,237 an asymmetric Friedel–Crafts reaction of a menthyl pyruvate ester238 and the first example of a highly diastereoselective hydrophosphorylation of a CN bond.239
2.5 Cycloadditions
Asymmetric cycloadditions provide an excellent method for the generation of cyclic structures with a number of stereogenic centres. The classical [4 + 2] cycloaddition of a diene with a dienophile may be conducted in an asymmetric manner by installing a stereodirecting group in either of these components. However, the most used approach is to incorporate the chiral auxiliary into the dienophile and the diastereoselectivity of such substrates with classically used auxiliaries such as sultams and hydrazides has been investigated as a function of solvent polarity.240 Applications of such reagents to synthesis include the use of chiral oxazolidinones in the preparation of the alkaloid (+)-gelsemine241 and in the synthesis of ceralure B.242 Extensive investigations of the asymmetric Diels–Alder additions of chiral sulfinyl benzoquinones with dienes have been carried out with and without added Lewis acid.243–245 Chiral sulfoxides based on naphthoquinones,246 sulfinyl and menthylsulfonyl furanones247 have also been employed. Brimble et al. have shown that pantolactone or phenylmenthyl auxiliaries exert poor stereocontrol (1.4 ∶ 1) on the cycloaddition of a naphthoquinone derivative and a diene.248 These auxiliaries also required destructive removal after the reaction was complete. Carbohydrate-derived acrylates based on isomannide249 and fructose250 have been employed with good levels of diastereoselectivity, and the origins of the selectivity investigated with the former. Formation of each diastereomer from the Diels–Alder addition of a chiral oxazoline dienophile with cyclopentadiene can be achieved by using either Et2AlCl or EtAlCl2.251 A chiral spirocyclic auxiliary 52 has been prepared via a lengthy procedure and used in asymmetric Diels–Alder reactions with excellent endo ∶ exo ratios and enantiomeric excess (Scheme 37).252 | ||
| Scheme 37 | ||
Diels–Alder additions of exocyclic methylenemorpholinones have been described including cyclopropanation and reduction, both with excellent levels of selectivity.253 A new chiral glycine anion equivalent has been disclosed derived from α-aminoisovalerophenone and been used in the preparation of cyclopropyl and norbornane α-amino acids with excellent enantioselectivity.254
In contrast to employing a stereodirecting group in the dienophile, surprisingly few reports have been made that use a stereodirecting group in the diene. Those that have been used include a chiral sulfoxide butadiene derivative 53255 and oxazolidinyl 54256 and glycosyl 55257,258 dienes (Scheme 38).
 | ||
| Scheme 38 | ||
A few examples also exist that use a tethered diene and dienophile or have used the auxiliary in both such components in the synthesis and include (−)-aminomenthol in the synthesis of tetrahydroepoxyisolindolines,259 (R)-2-phenylglycinol in the synthesis of solanoeclepin A,260 and menthol in the synthesis of kuehneromycin A.261
Hetero-Diels–Alder reactions provide the added advantage of the introduction of a heteroatom into the reaction product and can often result in excellent stereoselectivities. Use of a chiral amine as the auxiliary in the imine component of an aza-Diels–Alder reaction is a strategy that has been employed successfully. Bailey et al. reported such an addition of cyclopentadiene 57 with an imine derived from ethyl glyoxylate 56 with 90% asymmetric induction (Scheme 39).262 The [3.3.0] bicyclic systems 58 formed were subsequently converted to the N-oxides that spontaneously underwent a Meisenheimer-type rearrangement.
 | ||
| Scheme 39 | ||
A similar reaction has been performed with heterocyclic imines bearing the α-methylbenzylamine stereodirecting group in the presence of a Brønsted acid to give good yields and diastereoselectivity of the cycloadducts.263 A Lewis acid-catalysed Diels–Alder addition of a chiral imine and a diene has been reported with varying degrees of diastereoselectivity.264
Hetero-Diels–Alder reactions have also been performed using highly reactive chiral nitroso species. Stereodirecting groups such as chiral P-nitrosophosphates,265 carbohydrates,266,267 and pyroglutamates268 have been successfully used in this manner and, in the case of a nitroso sultam, employed in a formal synthesis of (−)-epibatidine.269,270
Several examples of an asymmetric Staudinger reaction have been reported that allow access to β-lactams, and auxiliaries that have been used in this reaction (with varying levels of stereocontrol) include chiral pyrrolidines,271 a new class of D-xylose-derived oxazolidinones,272 (+)-carene,273 and 2-phenylcyclohexanol.274 A double asymmetric [2 + 2] cycloaddition has also been carried out providing a single diastereomer in excellent yield.275 Asymmetric nitrile oxide cycloadditions have attracted considerable attention and the stereoselectivities of such intramolecular cyclisations have been investigated with a range of chiral auxiliaries.276 Only one report in this survey provided an example of chiral nitrile oxides from mannitol that reacted with 2-methylfuran with poor selectivity (3 ∶ 2).277 However, a number of examples where the chiral auxiliary has been installed in the acceptor have been reported. Magnesium ions were found to be essential for high diastereoselectivity in the nitrile oxide addition to chiral N-acryloyloxazolidinones278 and reactions with polymer-bound acceptors of this type have been investigated both in solution and with Merrifield and Wang grafted auxiliary.279 In general, the regio- and enantioselectivities were comparable but the solution yields were higher. Addition of nitrile oxides to camphor-derived auxiliaries 59 has been shown to occur with excellent diastereoselectivity in dichloromethane which dropped when THF was used as a solvent (Scheme 40).280
 | ||
| Scheme 40 | ||
Moving down one oxidation state from nitrile oxides, a number of examples of asymmetric addition of olefins to nitrones have been reported including using camphor derivatives in a formal synthesis of a carbapenem,281 the preparation of aza-2′,3′-dideoxynucleosides employing diastereoselective addition of a Vasella-type nitrone to vinyl acetate,282 using new morpholinone nitrones in the synthesis of carbocyclic polyoxin C,283 and in additions of furfural nitrones to an N-acryloyl chiral sultam.284 A chiral nitrone 60, itself prepared as a single diastereomer, has been used in intramolecular 1,3-dipolar cycloadditions with poor regioselectivity but excellent diastereoselectivity285 and this methodology applied to the synthesis of cylindricine-type alkaloids (Scheme 41).286
 | ||
| Scheme 41 | ||
Asymmetric nitrone cycloadditions have also been used in the synthesis of indolizidines using chiral nitrone287 and olefin acceptors.288 Denmark and co-workers described alternative routes to similar targets by making use of elegant tandem asymmetric [4 + 2]/[3 + 2] cyclisations giving products with good to excellent levels of diastereoselectivity that were used in the synthesis of (+)-casuarine, (−)-7-epiaustraline and (−)-1-epicastanospermine.289,290
Azomethine ylides have previously been used extensively in the facile preparation of polycyclic ring systems and novel amino acids. Intramolecular 1,3-dipolar cycloaddition of chiral morpholinone azomethine ylides with alkenes and alkynes has been investigated with semi-empirical calculations and the results agree with experimental observations.291 Intermolecular variants of this methodology have been used in the synthesis of syn- and anti-β -substituted α-amino acids292 and in the syntheses of (+)- and (−)-spirotryprostatin B.293 Diastereoselective cycloadditions of novel chiral azomethine ylide templates bearing two electronically different nitrogen substituents have also been reported.294 Finally, Carreira and co-workers have reported the asymmetric [3 + 2] cycloadditions of Me3SiCHN2 to unsaturated sultams in the synthesis of stelleamide A,295 and in the preparation of pyrazolines and aspartic acid derivatives.296
A number of other miscellaneous types of asymmetric cycloadditions deserve mentioning. Photolysis of the chromium carbene complexes 61 with the chiral ene carbamate 62 gave a good yield of the cyclobutanone 63 as a single diastereomer which was used in the preparation of (+)-aristeromycin and (+)-carbovir (Scheme 42).297
 | ||
| Scheme 42 | ||
In a similar manner, asymmetric [2 + 2] cycloaddition of a chiral chromium tricarbonyl imine with a ketene gave a β-lactam with excellent control of diastereoselectivity.298 Highly diastereoselective [2 + 2] cycloadditions have been performed using a chiral carbinol enol ether and dichloroketene with total regioselectivity and used in the synthesis of (−)-slaframine.299 β-Lactams have been prepared with complete diastereoselectivity by asymmetric [2 + 2] cycloaddition of a chiral oxazolidinone tethered ketene and subsequent asymmetric alkylation studies of the β-lactam investigated.300 Finally, the cycloaddition of a chiral cyclopropyl vinyl sulfoxide with acrylonitrile has been studied giving cyclopentyl derivatives with moderate levels of diastereoselectivity.301
2.6 Oxidation
In contrast to many of the other types of stoichiometric asymmetric processes that have been reported, examples of asymmetric oxidation processes are surprisingly scarce. As with asymmetric reduction, this is undoubtedly due to the highly successful catalytic asymmetric processes that now exist developed in particular by Jacobsen and Sharpless. Asymmetric epoxidation of proline-derived cinnamides 64 unexpectedly gave pyrrolidinooxazinones 65 and piperazinediones as the major products in good yield with an excellent ratio of diastereomers (Scheme 43).302 | ||
| Scheme 43 | ||
Dihydroxylation of unsaturated sultams has been shown to proceed with excellent diastereoselectivity for α,β substrates but poor selectivity for the β,γ analogues,303 while dihydroxylation of a chiral iron tricarbonyl compound has been used in the asymmetric synthesis of halicholactone.304 Finally, high levels of regio- and diastereoselectivity have been observed in the photooxygenation of chiral vinyl oxazolidines.305
2.7 Miscellaneous uses of chiral auxiliaries
Many natural products contain a cyclopropyl functionality and several methods have been explored for the introduction of such a group in a stereoselective manner, as exemplified in the synthesis of the glycolipids plakoside A and B,306 (−)-β-cuparenone and (−)-cuparane.307 Several other methods for asymmetric cyclopropanation deserve attention. Unsaturated chiral oxazolidinones have been converted to cyclopropanes using sulfur ylides in the presence of a Lewis acid with good to poor diastereoselectivities observed.308 The synthesis of enantiomerically pure cis configured cyclopropyl boronic esters has been achieved for the first time309 and similar methodology used to investigate the asymmetric cyclopropanation of alkenes bearing a borate ester auxiliary and a chiral substrate. In this case, the diastereoselectivity of those reactions using diazomethane were found to be auxiliary-controlled while the selectivity of Simmons–Smith cyclopropanation was controlled by the substrate.310 Bimetallic iron carbene–chromium tricarbonyl complexes have been used as substrates in the enantioselective synthesis of cyclopropanes giving enantioselectivities of >95% in some cases, the stereochemistry of which was controlled by the chromium moiety.311 Asymmetric cyclopropanation controlled by a chiral acetal 66 has been carried out using stable haloalkylzinc bipyridine derivatives 67 with excellent diastereomeric ratios obtained (Scheme 44).312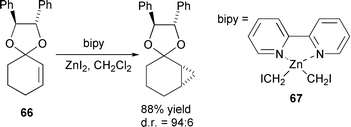 | ||
| Scheme 44 | ||
Asymmetric deprotonation, the stereochemistry of which is controlled by an adjacent stereodirecting group, is an excellent method for the selective remote introduction of functional groups. Asymmetric lithiation of an allyl carbamate induced by a complexing remote chiral group gave excellent diastereoselectivity after quenching with an electrophile, such as MeI.313 Directed metallation of arene transition metal complexes has been used successfully in many applications. Ortholithiation of an arene chromium tricarbonyl complex bearing a tartrate ligand followed by quenching with DMF proceeded in excellent diastereoselectivity (de >94%).314 Subsequent additions of organolithium and magnesium species to the aldehyde products of this reaction were investigated in some detail with diastereomeric excesses ranging from 82 to 94%. Directed metallation of a chromium tricarbonyl complex 68 and subsequent bromination controlled by an acetal stereodirecting group allowed the preparation of a key intermediate 69 in the synthesis of (−)-steganone (Scheme 45).315 Similiar methodology was used in the synthesis of korupensamine A316 and in an independent synthesis of (−)-steganone.317
 | ||
| Scheme 45 | ||
The synthesis of planar chiral ortho-functionalised ferrocenyl ketones has been achieved in good enantioselectivity (71–96%) using asymmetric deprotonation mediated by chiral hydrazones318 and O-methylephedrine.319 Similar methodology was used to prepare ferrocenes with planar and central chirality.320 Kagan has prepared enantiomerically pure ferrocene-based phosphines with only elements of planar chirality using asymmetric deprotonation mediated by chiral sulfoxides that can later be removed.321
Chiral enolates have not just been used in asymmetric alkylation and aldol reactions. Asymmetric amination α to a carbonyl has generated significant interest for use in amino acid synthesis. Potassium enolates of chiral oxazolidinones have been used successfully when reacted with trisyl azide (trisyl = 2,4,6-triisopropylbenzenesulfonyl) in the synthesis of an α-azidophosphotyrosyl mimic322 and in the synthesis of glycopeptide antibiotics related to teicoplanin.323 Such enolates bearing a β-stereogenic centre undergo highly diastereoselective aziridination with complete control of stereochemistry from the oxazolidinone stereogenic centre only.324 An interesting practical observation has been made in the quenching of such enolates with trisyl azide.325,326 Reproducible yields (75–95%) of the desired azides were obtained by addition of the solid trisyl azide in one portion at −78 °C. Electrophilic amination of enolates of chiral dithianes has been achieved with dibutyl azodicarboxylate giving relatively good levels of stereoselectivity, but with some loss of stereochemical integrity of the products that may have resulted during derivatisation procedures.327
Halogenation of chiral enolates is also of considerable synthetic importance. α-Fluoroalkylphosphonates have been prepared by quenching of the enolate of a chiral phosphonamide with N-fluorobenzenesulfonimide with poor selectivities.328 Davis et al. have used a highly diastereoselective fluorination of a dienolate of a chiral oxazolidinone in the synthesis of fluorinated carbohydrates.329 Other asymmetric halogenation reactions include stereoselective bromohydrin formation using glucose-derived auxiliaries to control the addition to a Michael acceptor,330 asymmetric bromination of acetals derived from tartrate derivatives,331 and an intramolecular bromoetherification of a chiral aldehyde to effect the kinetic resolution of norbornene-type substrates.332,333 Radical bromination and subsequent alcoholysis have also been investigated using a glucose-derived stereodirecting group giving good to poor levels of regio- and stereocontrol.334
Asymmetric ring opening reactions of chiral acetals and aza-acetals with nucleophiles has been performed with varying degrees of success. Allylsilanes have traditionally been used in nucleophilic additions to imines generated in situ and examples such as reaction with chiral bicyclic oxazolidines,335 chiral α-sulfonylalkylimidazolidones,336 and atropisomeric lactams337 have been reported. They have also been used in an intramolecular route to prepare piperidines,338 in the synthesis of (−)-coniceine339 and pipecolic acids.340 Lewis acid-mediated reaction of chiral silylketene and thioketene acetals with acetals and peroxyacetals has been studied with a number of auxiliaries, but poor selectivities have been observed.341
Enantiomerically pure C2 symmetric keto sulfoxides have been used to prepare symmetrical acetals 70 that undergo base-promoted asymmetric ring opening reactions giving the benzyl ether 71 in excellent yield and diastereomeric excess (Scheme 46).342,343
 | ||
| Scheme 46 | ||
Chiral oxazolidines derived from (R)-phenylglycinol react with dialkylalkynylalane–triethylamine complexes giving alkynylamines in excellent diastereomeric excess344 while enantiopure substituted pyrrolidines may be prepared by the reduction of chiral bicyclic oxazolidines with LiBHEt3.345
A number of miscellaneous reactions of organometallic reagents other than addition to carbonyl compounds have been reported. Cuprates have been shown to add to menthol derivatives in a highly diastereoselective SN2′ reaction, giving alkenes that can be cleaved by ozonolysis to chiral aldehydes.346,347 Alcohols from N-Boc oxazolidines have also been used in SN2′ organocuprate displacements with good stereoselectivities.348 Chiral α-chloro alkyl borates have been shown to undergo efficient SN2 displacement with Grignard reagents giving products of >99% ee.349 Chiral sulfoxides have been prepared by the sequential ring opening of chiral sulfites using organolithium and magnesium reagents.350 Stereocontrolled addition of Grignard reagents to chiral pyridinium salts facilitated access to tetrahydropyridines but with only modest levels of diastereoselectivity.351 Miscellaneous organometallic-based asymmetric transformations include the preparation of D-α-amino acids using a morpholinone template to control the stereochemistry of the condensation of a borate ester and aldehydes,352 the use of 8-phenylmenthol in samarium iodide-mediated radical formation–reprotonation,227 asymmetric Friedel–Crafts reaction,353 and a route to the synthesis of 11C enriched α-amino acids354 using lithium oxazolidinyl-derived auxiliaries. Reactive intermediates such as radicals and carbenoids have been used to effect stereoselective cyclisations. Such reactions of the former include using a new cyclohexanol chiral auxiliary to prepare chiral cyclopentanes but with low diastereoselectivity,355 in the asymmetric synthesis of (S)-pipecoline using reductive photocyclisation of dienamides controlled by a chiral amine,356 photoinduced electron transfer in the diastereoselective cyclisation of a prolinol derivative357 and in the synthesis of pyrrolidines.358,359 One example of a carbenoid-initiated cyclisation has been reported in the copper(II)-catalysed decomposition of a chiral diazomorpholinone that gave a 1 ∶ 2 mixture of diastereomeric bicyclic adducts.360
Stereoselective rearrangements provide an excellent method to introduce new stereogenic centres. Examples that have been reported include a highly diastereoselective thio-Claisen rearrangement using a C2 symmetric pyrrolidine stereodirecting group,361 desymmetrisation of a meso-N-hydroxyimide via a chiral Lossen rearrangement using a camphorsulfonyl auxiliary,362 asymmetric Meisenheimer rearrangements,363,364 an asymmetric Johnson ortho ester rearrangement of a chiral oxazolidine365 and a diastereoselective aza-Claisen rearrangement used in the synthesis of (−)-antimycin A3b.366
Stereoselective reductions of functional groups other than carbonyl groups have been carried out. Reduction of α,β-unsaturated sultams has been shown to give the highest levels of diastereoselectivity using hydrogenation conditions367 while the diastereoselective hydrogenation of ortho-substituted benzoic acid derivatives has been shown to be most effective using a pyroglutamic acid derivative.368 Asymmetric Birch reduction followed by quenching with methyl iodide has been optimised using proline-based auxiliaries 72 capable of controlling the intermediate enolate geometry by hindered rotation (Scheme 47).369 Products 73 with diastereomeric ratios of 93 ∶ 7 have been obtained in this way.
 | ||
| Scheme 47 | ||
A number of axially chiral biaryls have been prepared using a chiral template based upon L-threitol.370 BINOL has also been used as chiral tether for TiCl4-mediated oxidative coupling reactions giving chiral 2,3-diarylbutane-1,4-diols as single diastereomers.371 Axially chiral biaryls have also been prepared using diastereoselective cross coupling reactions and a series of such reactions have been carried out using chiral oxazolines.372 The selectivity was found to be dependent upon the structure of the substrate and selectivities of up to 8.5 ∶ 1 were obtained.
A number of other asymmetric transformations have been reported that cannot easily be grouped together. An asymmetric variant of a four component coupling of a silyl enol ether, chiral 1-alkoxy-1,3-diene, SO2 and an alkyl halide has been shown to occur with good to excellent diastereomeric excess, making use of a (S)-1-(2,4,6-triisopropylphenyl)ethyl stereodirecting group.373 A new chiral sulfenylating agent has been prepared and used in the sulfenylation of keto esters with good levels of enantioselectivity dependent upon the substrate structure374 and chiral diselenides have been used in the methoxy and hydroxyselenenylation of styrene giving good to excellent levels of diastereoselectivity.375 Chiral nucleophiles have been used in the asymmetric synthesis of β2-homoglycines by reaction of aryl ketenes with chiral alcohols, giving selectivities of greater than 90 ∶ 10376 and a similar transformation used in the first stages of a formal synthesis of roseophilin using menthol as the nucleophile, giving selectivities of 5 ∶ 1.377 Highly enantioselective propenoylation of lithium enolates of oxazolidinones has been carried out in the synthesis of the C13–C19 fragment of sanglifehrin A.378 Finally, diastereoselective ring opening reactions of gem-dibromocyclopropanes controlled by cyclohexyl and menthol derived auxiliaries have been used to prepare the core structures of epibatidine and anatoxin A.379
3. Chiral reagents
3.1 Chiral amine and lithium amide bases
Desymmetrisation of meso-ketones using chiral amines or lithium amide bases is now growing more widespread in use in organic synthesis. For example, the first step in the synthesis of the C38–C44 segment of altohyrtin A was asymmetric deprotonation with a chiral lithium amide, followed by silyl enol ether formation that proceeded in 95% enantiomeric excess.380 This methodology has been further extended by the use of new chiral lithium amides,381 chiral magnesium amides,382 in the synthesis of the pseudoguaiane framework,383 and chiral amine–silyl triflate complexes to generate chiral silyl enol ethers for a subsequent asymmetric tandem Michael–aldol reaction.384 Asymmetric deprotonation using chiral lithium amides 74 has provided substrates for subsequent asymmetric aldol reactions with good levels of diastereoselectivity (Scheme 48).385,386 | ||
| Scheme 48 | ||
Asymmetric deprotonation of epoxides can lead to unexpected rearrangement products and Kee et al. have disclosed preliminary observations of such reactions with cyclohexene epoxides.387 Attempted kinetic resolution of racemic epoxides with a chiral lithium amide base gave the product allylic alcohol (32% yield, 63% ee) and recovered starting material (51% yield, 44% ee). This method forms an excellent route for the preparation of highly functionalised cyclohexyl ring systems. Enantioselective deprotonation–rearrangement of meso-amino epoxides 75 gave good levels of stereoselectivity with a number of diamine-derived lithium amides 76 (Scheme 49).388
 | ||
| Scheme 49 | ||
The asymmetric deprotonation and subsequent quenching of substituted arene chromium tricarbonyl derivatives has been extensively studied with a variety of lithium amide bases and aryl directing groups.389 Axially chiral anilides have also been prepared with varying degrees of enantioselectivity by employing this methodology.390
3.2 Sparteine-mediated reactions and related processes
Interest in using sparteine and other chiral amines in mediating asymmetric deprotonation has been gaining momentum and this methodology appears to deliver excellent levels of stereoselectivity in many of the reactions studied. Beak et al. have reviewed similar chemistry to this, in particular, the use of dynamic thermodynamic resolution in achieving high levels of stereocontrol with particular emphasis on the use of chiral amines and organolithium species.391 Several interesting observations have been made in the area of (−)-sparteine-mediated deprotonation–alkylation, including those made by Deiters et al.392 Intramolecular trapping of an allylic anion in the presence of (−)-sparteine with an allylic chloride gave cyclonona-1,5-dienes in excellent enantiomeric excess. Introduction of an additional stereogenic centre in the substrate was found to only affect the diastereoselectivity of the reaction, not the enantioselectivity. A highly enantioselective anionic oxy-Cope rearrangement of the products was also shown to occur. A carbamate protecting group was used in this chemistry to direct metallation and a similar directed asymmetric deprotonation of a carbamate 77 in the presence of (−)-sparteine was followed by quenching with a variety of aldehydes 78. Excellent enantioselectivities were observed and the products used to make 5- to 8-membered ring unsaturated oxacycles (Scheme 50).393 | ||
| Scheme 50 | ||
Wiberg and Bailey have used molecular modeling to propose a transition state for the related enantioselective deprotonation of N-Boc pyrrolidine with isopropyllithium–(−)-sparteine.394 Carbamate groups are not essential for the directed metallation step and other methods have been used to generate the alkyllithium species. Addition of (−)-sparteine to aryllithiums prepared by lithium–halogen exchange facilitated the carbocyclic ring closure with a pendant allyl group giving selectivities of >86 ∶ 14 except when using THF as solvent.395 Identical work has also been reported with the additional feature that the resultant alkyllithium species 79 was trapped with a number of electrophiles (Scheme 51).396
 | ||
| Scheme 51 | ||
Nakamura et al. have demonstrated that α-thiobenzyllithium reagents react with aldehydes and ketones in the presence of chiral diamine ligands giving good yields and excellent levels of enantioselectivity.397 Of the diamines studied, bisoxazolines proved to be the most successful, while (−)-sparteine gave the worst results. Optically active phosphine–borane complexes have been prepared using (−)-sparteine–s-BuLi deprotonation followed by oxidation.398 The enantiomeric excess varied with the choice of alkyl group and with 1-adamantyl 93% was obtained. Sparteine-mediated deprotonation of medium ring epoxides has been investigated and gives different products with moderate to good levels of stereoselectivity depending upon the substrate structure and whether BF3·Et2O is added or not.399
Other chiral amines have also been used in similar reactions; chiral bisoxazolines have been used to induce chirality in the reaction of benzylic lithium reagents with benzophenone with good to excellent enantioselectivities.400 Similar studies have also been reported for methylation reactions.401 In a study of the catalytic use of a chiral amine in the asymmetric alkylation of achiral lithium enolates, stoichiometric use of these additives gave excellent yields and enantioselectivities using DME as solvent.402
Amine and amino alcohol additives have also been used successfully to control the stereochemistry of addition to carbonyl compounds. Diastereoselective additions of monosubstituted acetylenes to aldehydes have been shown to proceed in excellent enantioselectivity using N-methylephedrine and zinc triflate.403 These reactions have the advantage over similar dialkylzinc additions in that the reactions can be conducted in air. The asymmetric synthesis of (+)-salsolidine has been accomplished using the enantioselective addition of an alkyllithium to an imine in the presence of a chiral amino alcohol404 and a chiral C2 symmetric methyl ether 81 used to control the stereoselectivity of the addition of organolithium reagents to imines 80 with good to excellent enantioselectivities (Scheme 52).405
 | ||
| Scheme 52 | ||
Similar reactions have also been shown to occur in the presence of stoichiometric quantities of bisoxazoline ligands with different bite angles.406 Slight variations in enantioselectivities were observed with each of these ligands. The mechanism and structure of the intermediates used in the highly successful addition of lithium acetylides to an aryl trifluoromethyl ketone used in the asymmetric synthesis of efavirnez have been reported.407
3.3 Addition to CO, CN and CC bonds
Perhaps one of the most used chiral reagents for addition to CO bonds are the terpene-derived borane reagents originally developed by Brown and the origins of the stereoselectivity of B-chlorodiisopinocamphenylborane [(Ipc)2BCl] reagents have been investigated using molecular modeling.408 These reagents have been used to introduce the allyl functionality in a stereoselective manner and have been used in the total synthesis of many natural products including the macrocyclic core of apoptolidin,409 salicylihalamide A,107 epothilone analogues,410 lankacyclinol,411 lamoxirene,412 cryptophycin-24,413 the C29–C45 fragment of spongistatin 1,81 sanglifehrin A,414
δ-lactones,415 polyenes based upon (−)-stipiamide,416 nicotine analogues,417 and glycosphingolipids.418 Development of new methodology and applicability is still carried out in this field and Brown himself has reported application of this chemistry in the synthesis of both enantiomeric forms of perfluoroalkyl and aryl homoallylic alcohols.419 A new method for the preparation of chiral allylsilanes has also been reported using modified chiral allylborane reagents.420 Barrett et al. have described the synthesis of asymmetric bidirectional allylboration reagents 82 and their use in the preparation of C2 symmetric 1,5-diols 83 in excellent enantiomeric excess (Scheme 53).421 These diols were used to prepare enantiomerically pure spiroketals. | ||
| Scheme 53 | ||
Only one example of an asymmetric alkyl transfer reagent that uses a non-terpene stereodirecting group has been reported using the addition of an alkynyl BINOL borate to an unsaturated ketone with high regio- and stereoselectivities.422
Use of stoichiometric reducing agents is limited, undoubtedly for similar reasons as covered in Section 2.4. Binaphthol aluminium hydride complex (BINAL) has been used successfully to perform enantioselective reductions in the synthesis of (S)-coriolic acid423 and in the desymmetrisation of thioanhydrides used in the synthesis of d-biotin.424 Biomimetic NADH reagents have attracted some attention and macrocyclic analogues have been evaluated in the reduction of carbonyl compounds425 while chiral-bridged models have been used in the reduction of pyruvate analogues both with excellent enantioselectivities.426 Use of chiral dihydropyridine sulfoxides 84 in this sense has facilitated the asymmetric reduction–cyclisation of the ethylidenemalononitrile 85 with low levels of enantioselectivity (Scheme 54).427 Only one example in this survey of asymmetric hydroboration has been reported in the reduction of (E)- and (Z)-2-methoxybut-2-enes.428
 | ||
| Scheme 54 | ||
Chiral organometallic-based reagents have not been widely used, probably due to the difficulty in their preparation and/or expense. Of those described, chiral organotitanium reagents have been studied the most. The total synthesis of (+)-sedamine has been carried out using the key step of the enantioselective allylation of an aldehyde using a chiral titanium complex,429 methodology that has also been used in the synthesis of the lactone units of compactin and mevinolin.430 Allylation of acetylenic aldehydes with excellent enantiomeric excess has also been reported with this reagent.431 Chiral cyclopentadienyltitanium complexes based upon carbohydrates have been used in asymmetric aldol reactions giving the desired aldol products in good yield and enantiomeric excess (>90%).432 In the only example of a chiral allyltin reagent in this survey, addition of these reagents to N-acyliminium ions generated in situ proceeded with excellent levels of enantio- and diastereoselectivity.433 The matched/mismatching characteristics of this reaction were also evaluated.
Enantioselective aldol reactions have been carried out with chiral reagents, the most used of which appear to be chiral oxazaborolidine-mediated condensations with silyl ketene acetals and aldehydes434,435 and applied in the synthesis of amamistatin A436 and of a filipin III polyacetate unit.437 In the specific case of the addition of the silyl ketene acetal 86 to chiral aldehyde 87 in the presence of oxazaborolidine 88, excellent stereoselectivity was observed for the matched case and inverted stereoselection for the mismatched one (Scheme 55).438
 | ||
| Scheme 55 | ||
Corey and Choi have examined the use of chiral diazaborolidines in asymmetric aldol reactions and applied them to the synthesis of chloramphenicol.439,440 Chiral boron enolates have been prepared using (+)-(Ipc)2BCl and used to override the stereocontrolling elements of existing stereogenic centres in the synthesis of (+)-discodermolide.441
Several other transformations have been carried out on CO and CN bonds. Preparation of a cyclopropylstannane as a key intermediate in the synthesis of dictyopterene A was achieved in excellent enantioselectivity using the Charette asymmetric cycloprotonation methodology.442 Five molar equivalents of Zn(CH2I)2 were essential for high enantioselectivities. Chiral nucleophiles can be useful in deracemisation and desymmetrisation reactions. Thus, pantolactone has been used as a chiral nucleophile in the diastereoselective synthesis of α-phenyl δ-amino valeric acid with excellent diastereoselectivity by deracemisation of a ketene intermediate,443 while ring opening of meso-anhydrides with chiral carbinols gave good levels of diastereoselectivity but took 5 days for reaction to occur.444 Finally, chiral zirconium alkoxides have been used
to effect the enantioselective Meerwein–Ponndorf–Verley cyanation of aldehydes giving enantioselectivities of 61–91%.445
3.4 Miscellaneous uses of chiral reagents
A number of stoichiometric chiral agents have been used to promote cycloaddition reactions. Diels–Alder addition of butadiene to N-acryloyloxazolidinones proceeded in 85% yield and >92% ee in the presence of stoichiometric quantities of TADDOL ligands, products of which were used to prepare δ-peptide analogues of pyranosyl RNA.446 Significant improvements have been made in the selectivity of such reactions by addition of 4 Å molecular sieves.447 Chiral titanium(IV) complexes have also been used to promote Diels–Alder reactions of cyclohexa-1,3-diene and N-sulfinylbenzoyl carbamate with poor to good enantioselectivities.448 Other cycloadditions that have been carried out include a photochemical [2 + 2] cyclisation in the presence of a ‘chiral host’, the purpose of which was to participate in hydrogen bonding and transmit stereochemical information during the reaction, 449 and the use of chiral dioxazaborocines in nitrile oxide and nitrone cycloadditions.450The asymmetric Pauson–Khand reaction has attracted much attention and excellent selectivities have been obtained by preparing and using chiral dicobalt carbonyl reagents based on chiral propargylic alcohols.451 Chiral dicobalt carbonyl complexes bearing chiral ligands such as phosphines,452,453 and bidentate (P,S) ligands454 have also been employed generally with good selectivities. Optically pure heterobimetallic alkyne–cobalt complexes have also been used with excellent levels of enantioselectivity455 and brucine N-oxide has been employed as an ‘external’ source of chirality with encouraging enantioselectivities (Scheme 56).456
 | ||
| Scheme 56 | ||
A number of reactions involving organometallic species deserve attention; Coldham and Vennall have shown that the rate of cyclisation of chiral organolithium species is considerably slower for the formation of six-membered rings than for five-membered analogues.457 In the former case, this led to predominant racemisation giving products of low enantiomeric excess. Using a more reactive alkene acceptor increased the rate of the cyclisation giving improved enantiomeric excesses. The synthesis of a chiral secondary Grignard reagent has been achieved by making use of a carbenoid homologation procedure.458 Once prepared the reagent was found to be configurationally stable at −78 °C, racemising at higher temperatures. Addition of this reagent to phenyl isothiocyanate gave the addition product in excellent enantioselectivity, while oxidation gave secondary alcohols with varying degrees of selectivity. Copper-catalysed Grignard addition to alkene acceptors has been carried out using chiral dioxazaborocines as acceptors.459
Chiral organoaluminium reagents have been used to effect the stereoselective displacement of α-bromo amino acids using stoichiometric quantities of BINOL and trialkylaluminiums giving N-sulfonyl amino acids with moderate levels of enantioselectivity,460 while chiral aluminium trinaphthoxide derivatives have been used to effect a regio- and enantioselective siloxybutylation at the more hindered α-site of unsymmetrical ketones.461 Miscellaneous uses of organometallic reagents include addition of a Reformatsky-type reagent to a nitrone using a tartrate-derived additive 462 and one example of a titanium-mediated dihydrodimerisation of dihydrotetrahydrofurans in good enantiomeric excess using titanium TADDOLates.463
Asymmetric protonation of prochiral enolates has attracted much attention and the use of a wide range of chiral auxiliaries in enantioselective protonation has been studied.464 Lewis acid-assisted chiral Brønsted acids, such as those derived from BINOL and SnCl4, have been used successfully in enantioselective protonations of silyl enol ether and silyl ketene acetals both stoichiometrically and catalytically.465 Seebach and co-workers have reported the highly enantioselective protonation (>99% ee) of lithium enolates using TADDOL derivatives466 and a fluorous chiral proton source gave higher enantioselectivies than a non-fluorous analogue in the asymmetric protonation of a samarium enolate.467 Enantioselective protonation of an organolithium species was carried out using chiral amines with good degrees of selectivity (83–86% ee) and applied to the synthesis of salsolidine.468 Chiral bisphosphonates have been shown to adopt linear or cyclic macrocyclic structures when treated with HBF4.469 The former gave good levels of enantioselectivity when used in asymmetric protonation, while the latter demonstrated essentially no selectivity. Reasonable levels of enantioselectivity have also been observed by using chiral diamines.470 Enolates or their equivalents have also been shown to undergo asymmetric fluorination using chiral spirocyclic N-fluorosulfonamides471 and alkylation using a chiral alkylating agent used in the synthesis of (+)-chimonanthine.472 Several examples of asymmetric oxidants have been reported. Dichlorocamphorsulfonyloxaziridine has been shown to give significantly increased levels of diastereoselectivity (80 ∶ 20) compared to the parent oxaziridine in the asymmetric oxidation of an aromatic sulfide.473 Chiral hypervalent iodine oxidants 90 have been used in the oxidation of sulfides 91 with poor levels of enantioselectivity (Scheme 57).474
 | ||
| Scheme 57 | ||
Very few examples of cyclisation reactions that proceed under the influence of a chiral additive have been reported. Lewis acid-assisted chiral Brønsted acids, such as those derived from BINOL and SnCl4, have been used and have also been employed in enantioselective biomimetic cyclisations of isoprenoids with good to moderate levels of selectivity.475
Finally in this survey are a group of miscellaneous transformations that cannot easily be categorised elsewhere. A few examples of chiral organophosphorus chemistry have been reported. α-Methylbenzyl- and naphthylamines have been used in lanthanide three component couplings to prepare amino phosphonic acids with moderate diastereoselectivity.476 Enantioselective ring opening of meso-disulfides has been carried out using chiral phosphinamides with low levels of enantioselectivity.477 Phosphorothiolates containing a stereogenic phosphorus atom have been prepared with good selectivities (20 ∶ 1) using phosphorylating reagents derived from indole.478 Another asymmetric heteroatom involves the asymmetric amidoselenenylation of alkenes using a camphor-derived selenenylation reagent.479 Several radical or radical-type reactions have been performed. Inter- and intramolecular asymmetric radical carbon–carbon bond formation of sulfonamides has been studied using chiral diamine or TADDOL ligands with allyltributyltin reagents,480 while asymmetric pinacol coupling of aromatic aldehydes has been carried out in the presence of chiral amines and hydrazines giving diols with varying degrees of enantioselectivities.481 Chiral nucleophiles have been used in several applications. Use of either the sodium 91 or lithium alkoxide 92 of a chiral alcohol in the alcoholysis of a bis(vinyl sulfoxide) 93 leads to formation of either enantiomer of product 94 (Scheme 58).482
 | ||
| Scheme 58 | ||
Desymmetisation of meso-norbornadiene diesters with chiral alcohols and thiols gave poor to moderate degrees of diastereoselectivity,483 while an intramolecular oxy-Michael addition mediated by a carbohydrate derivative gave good levels of stereocontrol.484
Finally, an enantioselective Prins reaction has been developed using chiral tin BINOL derivatives that gives reasonable levels of enantioselectivity,485 and enantioselective acylating agents for amines have been described that perform excellently in kinetic resolutions of racemic piperidines.486
References
- B. Simoneau, P. Lavallée, M. Bailey, J.-S. Duceppe, C. Grand-Maître, L. Grenier, W. W. Ogilvie, M. A. Poupart and B. Thavonekham, Can. J. Chem., 2000, 78, 739 CrossRef CAS.
- M. P. Sibi, P. Liu and M. D. Johnson, Can. J. Chem., 2000, 78, 133 CrossRef CAS.
- M. D. Fletcher, J. R. Harding, R. A. Hughes, N. M. Kelly, H. Schmalz, A. Sutherland and C. L. Willis, J. Chem. Soc., Perkin Trans. 1, 2000, 43 RSC.
- J. R. Harding, R. A. Hughes, N. M. Kelly, A. Sutherland and C. L. Willis, J. Chem. Soc., Perkin Trans. 1, 2000, 3406 RSC.
- M. P. Sibi and P. K. Deshpande, J. Chem. Soc., Perkin Trans. 1, 2000, 1461 RSC.
- S. Ribe, R. K. Kondru, D. N. Beratan and P. Wipf, J. Am. Chem. Soc., 2000, 122, 4608 CrossRef CAS.
- A. G. H. Wee and Q. Yu, Tetrahedron Lett., 2000, 41, 587 CrossRef CAS.
- T. Tashiro, M. Bando and K. Mori, Synthesis, 2000, 1852 CrossRef CAS.
- M. T. Crimmins and K. A. Emmitte, Synthesis, 2000, 899 CrossRef CAS.
- T. Hasegawa and H. Yamamoto, Bull. Chem. Soc. Jpn., 2000, 73, 423 CrossRef CAS.
- N. J. S. Harmat, S. Mangani, E. Perrotta, D. Giannotti, R. Nannicini and M. Altamura, Tetrahedron Lett., 2000, 41, 1261 CrossRef CAS.
- H. Nakamura, K. Maruyama, K. Fujimaki and A. Murai, Tetrahedron Lett., 2000, 41, 1927 CrossRef CAS.
- J. Mulzer, G. Karig and P. Pojarliev, Tetrahedron Lett., 2000, 41, 7635 CrossRef CAS.
- M. Duan and L. A. Paquette, Tetrahedron Lett., 2000, 41, 3789 CrossRef CAS.
- R. Ponsinet, G. Chassaing, J. Vaissermann and S. Laville, Eur. J. Org. Chem., 2000, 83 CrossRef CAS.
- K. Mori, T. Tashiro and S. Sano, Tetrahedron Lett., 2000, 41, 5243 CrossRef CAS.
- D. C. Braddock and J. M. Brown, Tetrahedron: Asymmetry, 2000, 11, 3591 CrossRef CAS.
- N. Kise, T. Ueda, K. Kumada, Y. Terao and N. Ueda, J. Org. Chem., 2000, 65, 464 CrossRef CAS.
- M. P. Sibi and T. R. Rheault, J. Am. Chem. Soc., 2000, 122, 8873 CrossRef CAS.
- S. D. Bull, S. G. Davies, M.-S. Key, R. L. Nicholson and E. D. Savory, Chem. Commun., 2000, 1721 RSC.
- S. D. Bull, S. G. Davies, R. L. Nicholson, H. J. Sanganee and A. D. Smith, Tetrahedron: Asymmetry, 2000, 11, 3475 CrossRef CAS.
- S.-G. Lee, C. W. Lim, D. C. Kim and J. K. Lee, Tetrahedron: Asymmetry, 2000, 11, 1455 CrossRef CAS.
- A. A.-M. Abdel-Aziz, J. Okuno, S. Tanaka, T. Ishizuka, H. Matsunaga and T. Kunieda, Tetrahedron Lett., 2000, 41, 8533 CrossRef CAS.
- G. Guillena and C. Nájera, J. Org. Chem., 2000, 65, 7310 CrossRef CAS.
- L.-J. Gao, X.-Y. Zhao, M. Vandewalle and P. de Clercq, Eur. J. Org. Chem., 2000, 2755 CrossRef CAS.
- S. Hosokawa, K. Sekiguchi, M. Enemoto and S. Kobayashi, Tetrahedron Lett., 2000, 41, 6429 CrossRef CAS.
- A. Toró, P. Nowak and P. Deslongchamps, J. Am. Chem. Soc., 2000, 122, 4526 CrossRef CAS.
- D. Enders and A. Ridder, Synthesis, 2000, 1848 CrossRef CAS.
- D. Enders and C. Thiebes, Synlett, 2000, 1745 CAS.
- Z. Yang, A. B. Attygalle and J. Meinwald, Synthesis, 2000, 1936 CrossRef CAS.
- D. Enders, C. F. Janeck and G. Raabe, Eur. J. Org. Chem., 2000, 3337 CrossRef CAS.
- D. Enders, C. F. Janeck and J. Runsink, Synlett, 2000, 641 CAS.
- M. Fujita, O. Kitagawa, Y. Yamada, H. Izawa, H. Hasegawa and T. Taguchi, J. Org. Chem., 2000, 65, 1108 CrossRef CAS.
- J. Clayden, P. Johnson, J. H. Pink and M. Helliwell, J. Org. Chem., 2000, 65, 7033 CrossRef CAS.
- S. Pinheiro, C. B. S. S. Gonçalves, M. B. de Lima and F. M. C. de Farias, Tetrahedron: Asymmetry, 2000, 11, 3495 CrossRef CAS.
- Y. Matsukawa, J. S. D. Kumar, Y. Yoneyama, T. Katagiri and K. Uneyama, Tetrahedron: Asymmetry, 2000, 11, 4521 CrossRef CAS.
- M. Thieme, E. Vieira and J. Liebscher, Synthesis, 2000, 2051 CrossRef CAS.
- H. Pajouhesh, J. Chen and S. H. Pajouhesh, Tetrahedron: Asymmetry, 2000, 11, 4537 CrossRef CAS.
- S. Kojima, K. Fujitomo, Y. Shinohara, M. Shimizu and K. Ohkata, Tetrahedron Lett., 2000, 41, 9847 CrossRef CAS.
- P. D. Croce, C. L. Rosa and E. Pizzatti, Tetrahedron: Asymmetry, 2000, 11, 2635 CrossRef CAS.
- C. Ma, S. Yu, X. He, X. Liu and J. M. Cook, Tetrahedron Lett., 2000, 41, 2781 CrossRef CAS.
- B. Vivet, F. Cavelier and J. Martinez, Eur. J. Org. Chem., 2000, 807 CrossRef CAS.
- M. Adamczyk, S. R. Akireddy and R. E. Reddy, Tetrahedron, 2000, 56, 2379 CrossRef CAS.
- C. Nájera, T. Abellán and J. M. Sansano, Eur. J. Org. Chem., 2000, 2809 CrossRef CAS.
- W. Qiu, V. A. Soloshonok, C. Cai, X. Tang and V. J. Hruby, Tetrahedron, 2000, 56, 2577 CrossRef CAS.
- S. Collet, F. Carreaux, J.-L. Boucher, S. Pethe, M. Lepoivre, R. Danion-Bougot and D. Danion, J. Chem. Soc., Perkin Trans. 1, 2000, 177 RSC.
- X. Tang, V. A. Soloshonok and V. J. Hruby, Tetrahedron: Asymmetry, 2000, 11, 2917 CrossRef CAS.
- K. Miyashita, H. Miyabe, K. Tai, H. Iwaki and T. Imanishi, Tetrahedron, 2000, 56, 4691 CrossRef CAS.
- A. M. C. H. van den Nieuwendijk, N. M. A. J. Kriek, J. Brusse, J. H. van Boom and A. van der Gen, Eur. J. Org. Chem., 2000, 3683 CrossRef CAS.
- P. Kele, G. Sui, Q. Huo and R. M. Leblanc, Tetrahedron: Asymmetry, 2000, 11, 4959 CrossRef CAS.
- S. Chandrasekhar and A. Kausar, Tetrahedron: Asymmetry, 2000, 11, 2249 CrossRef CAS.
- K. W. Laue, S. Kröger, E. Wegelius and G. Haufe, Eur. J. Org. Chem., 2000, 3737 CrossRef CAS.
- F. Paradisi, G. Porzi, S. Rinaldi and S. Sandri, Tetrahedron: Asymmetry, 2000, 11, 4617 CrossRef CAS.
- F. Paradisi, G. Porzi, S. Rinaldi and S. Sandri, Tetrahedron: Asymmetry, 2000, 11, 1259 CrossRef CAS.
- S. E. Denmark and J.-H. Kim, Can. J. Chem., 2000, 78, 673 CrossRef CAS.
- S. Hanessian and L.-D. Cantin, Tetrahedron Lett., 2000, 41, 787 CrossRef CAS.
- L. P. Rapado, V. Bulugahapitiya and P. Renaud, Helv. Chim. Acta, 2000, 83, 1625 CrossRef CAS.
- H. Pajouhesh, M. Hosseini-Meresht, S. H. Pajouhesh and K. Curry, Tetrahedron: Asymmetry, 2000, 11, 4955 CrossRef CAS.
- M. Papke, S. Schulz, H. Tichy, E. Gingl and R. Ehn, Angew. Chem., Int. Ed., 2000, 39, 4339 CrossRef CAS.
- P. Laurent, J.-C. Braekman and D. Daloze, Eur. J. Org. Chem., 2000, 2057 CrossRef CAS.
- S. Kuwahara, J. Ishikawa, W. S. Leal, S. Hamade and O. Kodama, Synthesis, 2000, 1930 CrossRef CAS.
- K. Oda and A. I. Meyers, Tetrahedron Lett., 2000, 41, 8193 CrossRef CAS.
- J. Lin, W. H. Chan, A. W. M. Lee, W. Y. Wong and P. Q. Huang, Tetrahedron Lett., 2000, 41, 2949 CrossRef CAS.
- J. D. Dudones and A. J. Pearson, Tetrahedron Lett., 2000, 41, 8037 CrossRef CAS.
- S. Huang and D. L. Comins, Chem. Commun., 2000, 569 RSC.
- D. B. Berkowitz, J. M. McFadden and M. K. Sloss, J. Org. Chem., 2000, 65, 2907 CrossRef CAS.
- J. Mulzer, O. Langer, M. Hiersemann, J. W. Bats, J. Buschmann and P. Luger, J. Org. Chem., 2000, 65, 6540 CrossRef CAS.
- J. H. Bailey, A. T. J. Byfield, P. J. Davis, A. C. Foster, M. Leech, M. G. Moloney, M. Müller and C. K. Prout, J. Chem. Soc., Perkin Trans. 1, 2000, 1977 RSC.
- S. M. Allin, D. G. Vaidya, M. I. Page, A. M. Z. Slawin and T. Smith, Tetrahedron Lett., 2000, 41, 2219 CrossRef CAS.
- J. L. Vicario, D. Badía, E. Domínguez and L. Carrillo, Tetrahedron: Asymmetry, 2000, 11, 3779 CrossRef CAS.
- V. Jullian, J.-C. Quirion and H.-P. Husson, Eur. J. Org. Chem., 2000, 1319 CrossRef CAS.
- D. Yang, X.-Y. Ye and M. Xu, J. Org. Chem., 2000, 65, 2208 CrossRef CAS.
- M. Tada, S. Nishiiri, Y. Zhixiang, Y. Imai, S. Tajima, N. Okazaki, Y. Kitano and K. Chiba, J. Chem. Soc., Perkin Trans. 1, 2000, 2657 RSC.
- A. Haudrechy, C. Chassaing, C. Riche and Y. Langlois, Tetrahedron, 2000, 56, 3181 CrossRef CAS.
- D.-H. Lee and M.-D. Rho, Tetrahedron Lett., 2000, 41, 2573 CrossRef CAS.
- V. Alezra, M. Bonin, A. Chiaroni, L. Micouin, C. Riche and H.-P. Husson, Tetrahedron Lett., 2000, 41, 1737 CrossRef CAS.
- V. Alezra, M. Bonin, L. Micouin and H.-P. Husson, Tetrahedron Lett., 2000, 41, 651 CrossRef CAS.
- K.-H. Baringhaus, H. Matter and M. Kurz, J. Org. Chem., 2000, 65, 5031 CrossRef CAS.
- M. Christmann, U. Bhatt, M. Quitschalle, E. Claus and M. Kalesse, Angew. Chem., Int. Ed., 2000, 39, 4364 CrossRef CAS.
- S. Lemaire-Audoire and P. Vogel, J. Org. Chem., 2000, 65, 3346 CrossRef CAS.
- G. C. Micalizio, A. N. Pinchuk and W. R. Roush, J. Org. Chem., 2000, 65, 8730 CrossRef CAS.
- G. A. Wallace, R. W. Scott and C. H. Heathcock, J. Org. Chem., 2000, 65, 4145 CrossRef CAS.
- M. G. N. Russell, R. Baker, R. G. Ball, S. R. Thomas, N. N. Tsou and J. L. Castro, J. Chem. Soc., Perkin Trans. 1, 2000, 893 RSC.
- A. N. Hulme, C. H. Montgomery and D. K. Henderson, J. Chem. Soc., Perkin Trans. 1, 2000, 1837 RSC.
- F. Yokokawa, H. Fujiwara and T. Shioiri, Tetrahedron, 2000, 56, 1759 CrossRef CAS.
- R. J. Davenport and A. C. Regan, Tetrahedron Lett., 2000, 41, 7619 CrossRef CAS.
- C. Hermann, G. C. G. Pais, A. Geyer, S. M. Kühnert and M. E. Maier, Tetrahedron, 2000, 56, 8461 CrossRef CAS.
- T. Sunazuka, T. Hirose, T. Shirahata, Y. Harigaya, M. Hayashi, K. Komiyama, S. Omura and A. B. Smith, J. Am. Chem. Soc., 2000, 122, 2122 CrossRef CAS.
- N. Waizumi, T. Itoh and T. Fukuyama, J. Am. Chem. Soc., 2000, 122, 7825 CrossRef CAS.
- A. B. Smith, T. J. Beauchamp, M. J. LaMarche, M. D. Kaufman, Y. P. Qiu, H. Arimoto, D. R. Jones and K. Kobayashi, J. Am. Chem. Soc., 2000, 122, 8654 CrossRef.
- J. Eames, D. J. Fox, M. A. de las Heras and S. Warren, J. Chem. Soc., Perkin Trans. 1, 2000, 1903 RSC.
- S. K. Chattopadhyay and G. Pattenden, J. Chem. Soc., Perkin Trans. 1, 2000, 2429 RSC.
- G. J. McGarvey, J. A. Mathys and K. J. Wilson, Tetrahedron Lett., 2000, 41, 6011 CrossRef CAS.
- R. Kuang, A. K. Ganguly, T.-M. Chan, B. N. Pramanik, D. J. Blythin, A. T. McPhail and A. K. Saksena, Tetrahedron Lett., 2000, 41, 9575 CrossRef CAS.
- D. A. Evans, V. J. Cee, T. E. Smith, D. M. Fitch and P. S. Cho, Angew. Chem., Int. Ed., 2000, 39, 2533 CrossRef CAS.
- Z. Li, R. Wu, R. Michalczyk, R. B. Dunlap, J. D. Odom and L. A. Silks, J. Am. Chem. Soc., 2000, 122, 386 CrossRef CAS.
- M. T. Crimmins and E. A. Tabet, J. Am. Chem. Soc., 2000, 122, 5473 CrossRef CAS.
- M. T. Crimmins, B. W. King, W. J. Zuercher and A. L. Choy, J. Org. Chem., 2000, 65, 8499 CrossRef CAS.
- M. A. Brimble, M. R. Nairn and J. S. O. Park, J. Chem. Soc., Perkin Trans. 1, 2000, 697 RSC.
- B. Panicker, J. M. Karle and M. A. Avery, Tetrahedron, 2000, 56, 7859 CrossRef CAS.
- I. Efremov and L. A. Paquette, J. Am. Chem. Soc., 2000, 122, 9324 CrossRef CAS.
- S. Fukuzawa, H. Matsuzawa and S. Yoshimitsu, J. Org. Chem., 2000, 65, 1702 CrossRef CAS.
- M. Stöver, A. Lützen and P. Köll, Tetrahedron: Asymmetry, 2000, 11, 371 CrossRef CAS.
- T. H. Kim and G. J. Lee, Tetrahedron Lett., 2000, 41, 1505 CrossRef CAS.
- S. Caddick, N. J. Parr and M. C. Pritchard, Tetrahedron Lett., 2000, 41, 5963 CrossRef CAS.
- P. Perlmutter and W. Selajarern, Aust. J. Chem., 2000, 53, 349 CrossRef CAS.
- Y. Wu, L. Esser and J. K. De Brabander, Angew. Chem., Int. Ed., 2000, 39, 4308 CrossRef CAS.
- J. L. Vicario, D. Badía, E. Domínguez, M. Rodríguez and L. Carrillo, J. Org. Chem., 2000, 65, 3754 CrossRef CAS.
- J. L. Vicario, D. Badiá, E. Domínguez, M. Rodríguez and L. Carrillo, Tetrahedron Lett., 2000, 41, 8297 CrossRef CAS.
- E. Hedenström, F. Andersson and M. Hjalmarsson, J. Chem. Soc., Perkin Trans. 1, 2000, 1513 RSC.
- I. Paterson, V. A. Doughty, M. D. McLeod and T. Trieselmann, Angew. Chem., Int. Ed., 2000, 39, 1308 CrossRef CAS.
- D. Suzuki, H. Urabe and F. Sato, Angew. Chem., Int. Ed., 2000, 39, 3290 CrossRef CAS.
- C. Palomo, M. Oiarbide, A. K. Sharma, M. C. González-Rego, A. Linden, J. M. Garćia and A. González, J. Org. Chem., 2000, 65, 9007 CrossRef CAS.
- A. Solladié-Cavallo and B. Crescenzi, Synlett, 2000, 327 CAS.
- P. T. Grover, N. N. Bhongle, S. A. Wald and C. H. Senanayake, J. Org. Chem., 2000, 65, 6283 CrossRef CAS.
- S. Usse, G. Guillaumet and M.-C Viaud, J. Org. Chem., 2000, 65, 914 CrossRef CAS.
- S. Florio, V. Capriati and R. Luisi, Tetrahedron Lett., 2000, 41, 5295 CrossRef CAS.
- A. G. Waterson and A. I. Meyers, J. Org. Chem., 2000, 65, 7240 CrossRef CAS.
- Y. Arai, T. Masuda, S. Yoneda, Y. Masaki and M. Shiro, J. Org. Chem., 2000, 65, 258 CrossRef CAS.
- D. Zhang, C. Bleasdale, B. T. Golding and W. P. Watson, Chem. Commun., 2000, 1141 RSC.
- J. L. G. Ruano, D. Barros, M. C. Maestro, A. M. Z. Slawin and P. C. B. Page, J. Org. Chem., 2000, 65, 6027 CrossRef CAS.
- M. Toyota, T. Wada and M. Ihara, J. Org. Chem., 2000, 65, 4565 CrossRef CAS.
- J. Christoffers and A. Mann, Angew. Chem., Int. Ed., 2000, 39, 2752 CrossRef CAS.
- M. Amat, J. Bosch, J. Hidalgo, M. Cantó, M. Pérez, N. Llor, E. Molins, C. Miravitlles, M. Orozco and J. Luque, J. Org. Chem., 2000, 65, 3074 CrossRef CAS.
- C. Schneider and O. Reese, Synthesis, 2000, 1689 CrossRef CAS.
- M. Wu, T. Okino, L. M. Nogle, B. L. Marquez, R. T. Williamson, N. Sitachitta, F. W. Berman, T. F. Murray, K. McGough, R. Jacobs, K. Colsen, T. Asano, F. Yokokawa, T. Shioiri and W. H. Gerwick, J. Am. Chem. Soc., 2000, 122, 12041 CrossRef CAS.
- S. Wang, X. Tang and V. J. Hruby, Tetrahedron Lett., 2000, 41, 1307 CrossRef CAS.
- D. R. Williams, P. D. Lowder and Y.-G. Gu, Tetrahedron Lett., 2000, 41, 9397 CrossRef CAS.
- C. Scheider and O. Reese, Angew. Chem., Int. Ed., 2000, 39, 2948 CrossRef.
- T. Tomoyasu, K. Tomooka and T. Nakai, Tetrahedron Lett., 2000, 41, 345 CrossRef CAS.
- K. Afarinkia, H. M. Binch and I. Forristal, Synlett, 2000, 1771 CAS.
- M. Bella, R. Margarita, C. Orlando, M. Orsini, L. Parlanti and G. Piancatelli, Tetrahedron Lett., 2000, 41, 561 CrossRef CAS.
- S. V. Kolotuchin and A. I. Meyers, J. Org. Chem., 2000, 65, 3018 Search PubMed.
- M. Mitsuya, Y. Ogino, N. Ohtake and T. Mase, Tetrahedron, 2000, 56, 9901 CrossRef CAS.
- R. Aslanian, G. Lee, R. V. Iyer, N. Y. Shih, J. J. Piwinski, R. W. Draper and A. T. McPhail, Tetrahedron: Asymmetry, 2000, 11, 3867 Search PubMed.
- J. H. Bailey, D. T. Cherry, J. Dyer, M. G. Moloney, M. J. Bamford, S. Keeling and R. B. Lamont, J. Chem. Soc., Perkin Trans. 1, 2000, 2783 RSC.
- K. Takasu, M. Ueno and M. Ihara, Tetrahedron Lett., 2000, 41, 2145 CrossRef CAS.
- A. Ali, V. U. Ahmad, B. Ziemer and J. Liebscher, Tetrahedron: Asymmetry, 2000, 11, 4365 CrossRef CAS.
- V. A. Soloshonok, C. Cai and V. J. Hruby, Angew. Chem., Int. Ed., 2000, 39, 2172 CrossRef CAS.
- V. A. Soloshonok, C. Cai and V. J. Hruby, Tetrahedron Lett., 2000, 41, 9645 CrossRef CAS.
- M. Nour, K. Tan, C. Cavé, D. Villeneuve, D. Desmaële, J. d'Angelo and C. Riche, Tetrahedron: Asymmetry, 2000, 11, 995 CrossRef CAS.
- G. Revial, I. Jabin and M. Pfau, Tetrahedron: Asymmetry, 2000, 11, 4975 Search PubMed.
- D. Enders, P. Teschner and G. Raabe, Synlett, 200, 637 Search PubMed.
- V. Ojea, M. Ruiz, G. Shapiro and E. Pombo-Villar, J. Org. Chem., 2000, 65, 1984 CrossRef CAS.
- C. Baldoli, S. Maiorana, E. Licandro, D. Perdicchia and B. Vandoni, Tetrahedron: Asymmetry, 2000, 11, 2007 CrossRef CAS.
- D. François, E. Poupon, M.-C. Lallemand, N. Kunesch and H.-P. Husson, J. Org. Chem., 2000, 65, 3209 CrossRef CAS.
- G. Revial, S. Lim, B. Viossat, P. Lemoine, A. Tomas, A. F. Duprat and M. Pfau, J. Org. Chem., 2000, 65, 4593 CrossRef CAS.
- E. Licandro, S. Maiorana, C. Baldoli, L. Capella and D. Perdicchia, Tetrahedron: Asymmetry, 2000, 11, 975 CrossRef CAS.
- S. Nakamura, Y. Watanabe and T. Toru, J. Org. Chem., 2000, 65, 1758 CrossRef CAS.
- I. Fernńdez, C. S. Araújo, M. J. Romero, F. Alcudia and N. Khiar, Tetrahedron, 2000, 56, 3749 CrossRef.
- S. Karlsson and H.-E. Högberg, Synthesis, 2000, 1863 CrossRef CAS.
- S. Hanessian, A. Gomtsyan and N. Malek, J. Org. Chem., 2000, 65, 5623 CrossRef CAS.
- M. Marinozzi and R. Pellicciari, Tetrahedron Lett., 2000, 41, 9125 CrossRef CAS.
- N. Mase, Y. Watanabe, T. Toru, T. Kakumoto and T. Hagiwara, J. Org. Chem., 2000, 65, 7083 CrossRef CAS.
- R. Munakata, K. Totani, K. Takao and K. Tadano, Synlett, 2000, 979 CAS.
- G. Campari, M. Fagnoni, M. Mella and A. Albini, Tetrahedron: Asymmetry, 2000, 11, 1891 CrossRef CAS.
- S. G. Davies, C. A. P. Smethurst, A. D. Smith and G. D. Smyth, Tetrahedron: Asymmetry, 2000, 11, 2437 CrossRef CAS.
- S. D. Bull, S. G. Davies, G. Fenton, A. W. Mulvaney, R. S. Prasad and A. D. Smith, J. Chem. Soc., Perkin Trans. 1, 2000, 3765 RSC.
- S. D. Bull, S. G. Davies, S. Delgado-Ballester, G. Fenton, P. M. Kelly and A. D. Smith, Synlett, 2000, 1257 CAS.
- P. O'Brien, D. W. Porter and N. M. Smith, Synlett, 2000, 1336 CAS.
- M. Baenziger, L. Gobbi, B. P. Riss, F. Schaefer and A. Vaupel, Tetrahedron: Asymmetry, 2000, 11, 2231 CrossRef CAS.
- D. Ma and H. Sun, Tetrahedron Lett., 2000, 41, 1947 CrossRef CAS.
- D. Ma and H. Sun, J. Org. Chem., 2000, 65, 6009 CrossRef CAS.
- D. Enders, S. F. Müller, G. Raabe and J. Runsink, Eur. J. Org. Chem., 2000, 879 CrossRef CAS.
- D. Enders, J. Vázquez and G. Raabe, Eur. J. Org. Chem., 2000, 893 CrossRef CAS.
- D. Lucet, P. Heyse, A. Gissot, T. Le Gall and C. Mioskowski, Eur. J. Org. Chem., 2000, 3575 CrossRef CAS.
- A. Volonterio, P. Bravo, N. Moussier and M. Zanda, Tetrahedron Lett., 2000, 41, 6517 CrossRef CAS.
- G. Cardillo, L. Gentilucci, M. Gianotti and A. Tolomelli, Eur. J. Org. Chem., 2000, 2489 CrossRef CAS.
- C. P. Rayner (née Baird), A. J. Clark, S. M. Rooke, T. J. Sparey and P. C. Taylor, J. Chem. Soc., Perkin Trans. 1, 2000, 79 RSC.
- L. A. Paquette, J. Tae, M. P. Arrington and A. H. Sadoun, J. Am. Chem. Soc., 2000, 122, 2742 CrossRef CAS.
- N. A. Braun, M. Ousmer, J. D. Bray, D. Bouchu, K. Peters, E.-M. Peters and M. A. Ciufolini, J. Org. Chem., 2000, 65, 4397 CrossRef CAS.
- M. Node, K. Nishide, Y. Shigeta, H. Shiraki and K. Obata, J. Am. Chem. Soc., 2000, 122, 1927 CrossRef CAS.
- H. Shiraki, K. Nishide and M. Node, Tetrahedron Lett., 2000, 41, 3437 CrossRef CAS.
- C.-H. Lin, K.-S. Yang, J.-F. Pan and K. Chen, Tetrahedron Lett., 2000, 41, 6815 CrossRef CAS.
- A. Ali, V. U. Ahmad, J. Leistner and J. Liebscher, J. Chem. Soc., Perkin. Trans. 1, 2000, 1897 RSC.
- D. Enders, L. Tedeschi and J. W. Bats, Angew. Chem., Int. Ed., 2000, 39, 4605 CrossRef CAS.
- A. P. Degnan and A. I. Meyers, J. Org. Chem., 2000, 65, 3503 CrossRef CAS.
- Y. Matsumura, Y. Kanda, K. Shirai, O. Onomura and T. Maki, Tetrahedron, 2000, 56, 7411 CrossRef CAS.
- T. Kawakami, H. Ohtake, H. Arakawa, T. Okachi, Y. Imada and S.-I. Murahashi, Bull. Chem. Soc. Jpn., 2000, 73, 2423 CrossRef CAS.
- A. B. McLaren and J. B. Sweeney, Synlett, 2000, 1625 CAS.
- C. Palomo, M. Oiarbide, M. C. González-Rego, A. K. Sharma, J. M. García, A. González, C. Landa and A. Linden, Angew. Chem., Int. Ed., 2000, 39, 1063 CrossRef CAS.
- F. A. Davis, T. Fang, B. Chao and D. M. Burns, Synthesis, 2000, 2106 CrossRef CAS.
- F. Steif, B. Wibbeling, O. Meyer and D. Hoppe, Synthesis, 2000, 743 CrossRef CAS.
- A. Kreier, R. Fröhlich, E. Wegelius and D. Hoppe, Synthesis, 2000, 1391 CrossRef CAS.
- B. Dudot, J. Royer, M. Sevrin and P. George, Tetrahedron Lett., 2000, 41, 4367 CrossRef CAS.
- B. Dudot, A. Chiaroni and J. Royer, Tetrahedron Lett., 2000, 41, 6355 CrossRef CAS.
- P. Allef and H. Kunz, Tetrahedron: Asymmetry, 2000, 11, 375 CrossRef CAS.
- J. L. G. Ruano, M. C. Carreño, M. A. Toledo, J. M. Aguirre, M. T. Aranda and J. Fischer, Angew. Chem., Int. Ed., 2000, 39, 2736 CrossRef CAS.
- J. L. G. Ruano, A. Alcudia, M. del Prado, D. Barros, M. C. Maestro and I. Fernández, J. Org. Chem., 2000, 65, 2856 CrossRef CAS.
- C. A. Brooks and D. L. Comins, Tetrahedron Lett., 2000, 41, 3551 CrossRef CAS.
- C. Agami, F. Couty and G. Evano, Tetrahedron: Asymmetry, 2000, 11, 4639 CrossRef CAS.
- N. A. Magnus, P. N. Confalone and L. Storace, Tetrahedron Lett., 2000, 41, 3015 CrossRef CAS.
- A. Giuseppe, G. Alvaro, F. Grepioni, S. Grilli, L. Maini, G. Martelli and D. Savoia, Synthesis, 2000, 581.
- S. Roland and P. Mangeney, Eur. J. Org. Chem., 2000, 611 CrossRef CAS.
- T. Yamauchi, K. Higashiyama, H. Kubo and S. Ohmiya, Tetrahedron: Asymmetry, 2000, 11, 3003 CrossRef CAS.
- W. H. Chan, A. W. M. Lee, P. F. Xia and W. Y. Wong, Tetrahedron Lett., 2000, 41, 5725 CrossRef CAS.
- S. Nakamura, H. Yasuda, Y. Watanabe and T. Toru, Tetrahedron Lett., 2000, 41, 4157 CrossRef CAS.
- S. Nakamura, H. Yasuda, Y. Watanabe and T. Toru, J. Org. Chem., 2000, 65, 8640 CrossRef CAS.
- A. Ichikawa, S. Hiradate, A. Sugio, S. Kuwahara, M. Watanabe and N. Harada, Tetrahedron: Asymmetry, 2000, 11, 2669 CrossRef CAS.
- T. Fukuda, A. Takehara, N. Haniu and M. Iwao, Tetrahedron: Asymmetry, 2000, 11, 4083 CrossRef CAS.
- G. Martelli, S. Morri and D. Savoia, Tetrahedron, 2000, 56, 8367 CrossRef CAS.
- C. Agami, S. Comesse and C. Kadouri-Puchot, Tetrahedron Lett., 2000, 41, 6059 CrossRef CAS.
- J. Clayden, C. McCarthy and J. G. Cumming, Tetrahedron Lett., 2000, 41, 3279 CrossRef CAS.
- D. Enders and C. Thiebes, Synthesis, 2000, 510 CrossRef CAS.
- D. Enders and J. H. Kirchhoff, Synthesis, 2000, 2099 CrossRef CAS.
- R. E. Gawley, Q. Zhang and A. T. McPhail, Tetrahedron: Asymmetry, 2000, 11, 2093 CrossRef CAS.
- R. Kumareswaran and T. Balasubramanian, Tetrahedron Lett., 2000, 41, 8157 CrossRef CAS.
- A. Alexakis, A. Tomassini, C. Chouillet, S. Roland, P. Mangeney and G. Bernardinelli, Angew. Chem., Int. Ed., 2000, 39, 4093 CrossRef CAS.
- T. Hamada, R. Mizojiri, H. Urabe and F. Sato, J. Am. Chem. Soc., 2000, 122, 7138 CrossRef CAS.
- S. Okamoto, K. Fukuhara and F. Sato, Tetrahedron Lett., 2000, 41, 5561 CrossRef CAS.
- T.-P. Loh, J.-M. Huang, K.-C. Xu, S.-H. Goh and J. J. Vittal, Tetrahedron Lett., 2000, 41, 6511 CrossRef CAS.
- C. Cimarelli and G. Palmieri, Tetrahedron: Asymmetry, 2000, 11, 2555 Search PubMed.
- E. M. Cayuela, L. Xiao, T. Sturm, B. R. Manzano, F. A. Jalon and W. Weissensteiner, Tetrahedron: Asymmetry, 2000, 11, 861 CrossRef CAS.
- F. Segat-Dioury, O. Lingibé, B. Graffe, M.-C. Sacquet and G. Lhommet, Tetrahedron, 2000, 56, 233 CrossRef CAS.
- S. Cutri, M. Bonin, L. Micouin, O. Froelich, J.-C. Quirion and H.-P. Husson, Tetrahedron Lett., 2000, 41, 1179 CrossRef CAS.
- P. Bataille, M. Paterne and E. Brown, Tetrahedron: Asymmetry, 2000, 11, 1165 CrossRef CAS.
- S. Nakamura, M. Kuroyanagi, Y. Watanabe and T. Toru, J. Chem. Soc., Perkin Trans. 1, 2000, 3143 RSC.
- G. Solladié, X. J. Salom-Roig and G. Hanquet, Tetrahedron Lett., 2000, 41, 551 CrossRef CAS.
- F. Yuste, B. Ortiz, A. Carrasco, M. Peralta, L. Qunitero, R. Sánchez-Obregón, F. Walls and J. L. G. Ruano, Tetrahedron: Asymmetry, 2000, 11, 3079 CrossRef CAS.
- G. Solladié, L. Gressot and F. Colobert, Eur. J. Org. Chem., 2000, 357 CrossRef CAS.
- C. S. Park, H. G. Choi, H. Lee, W. K. Lee and H.-J. Ha, Tetrahedron: Asymmetry, 2000, 11, 3283 CrossRef CAS.
- P. de March, M. Escoda, M. Figueredo, J. Font, E. García-García and S. Rodríguez, Tetrahedron: Asymmetry, 2000, 11, 4473 CrossRef CAS.
- G. K. Friestad and J. Qin, J. Am. Chem. Soc., 2000, 122, 8329 CrossRef CAS.
- H. Miyabe, C. Ushiro, M. Ueda, K. Yamakawa and T. Naito, J. Org. Chem., 2000, 65, 176 CrossRef CAS.
- M. P. Bertrand, S. Coantic, L. Feray, R. Nouguier and P. Perfetti, Tetrahedron, 2000, 56, 3951 CrossRef CAS.
- S. M. Kim, I. S. Byun and Y. H. Kim, Angew. Chem., Int. Ed., 2000, 39, 728 CrossRef CAS.
- J.-M. Fang, M.-Y. Chen, J.-S. Shiue, L. Lu and J.-L. Hsu, Tetrahedron Lett., 2000, 41, 4633 CrossRef CAS.
- S. Iwama, W.-G. Gao, T. Shinada and Y. Ohfune, Synlett, 2000, 1631 CAS.
- F. A. Davis, V. Srirajan, D. L. Fanelli and P. Portonovo, J. Org. Chem., 2000, 65, 7663 CrossRef CAS.
- F. A Davis, S. Lee, H. Zhang and D. L. Fanelli, J. Org. Chem., 2000, 65, 8704 CrossRef CAS.
- F. A. Davis and V. Srirajan, J. Org. Chem., 2000, 65, 3248 CrossRef CAS.
- S. P. Vincent, A. Schleyer and C.-H. Wong, J. Org. Chem., 2000, 65, 4440 CrossRef CAS.
- J. Wede, F.-J. Volk and A. W. Frahm, Tetrahedron: Asymmetry, 2000, 11, 3231 CrossRef CAS.
- W.-W. Huang, P. A. Moore, A. R. Far and T. T. Tidwell, J. Org. Chem., 2000, 65, 3877 CrossRef CAS.
- M. Marzi, P. Minetti, G. Moretti, M. O. Tinti and F. De Angelis, J. Org. Chem., 2000, 65, 6766 CrossRef CAS.
- D. J. Dixon, S. V. Ley and D. J. Reynolds, Angew. Chem., Int. Ed., 2000, 39, 3622 CrossRef CAS.
- A. Solladié-Cavallo, L. Bouérat and M. Roje, Tetrahedron Lett., 2000, 41, 7309 CrossRef CAS.
- F. M. Hauser and D. Ganguly, J. Org. Chem., 2000, 65, 1842 CrossRef CAS.
- I. Schlemminger, A. Lützen, A. Willecke, W. Maison, R. Koch, W. Saak and J. Martens, Tetrahedron Lett., 2000, 41, 7285 CrossRef CAS.
- C. Chapuis, A. Kucharska and J. Jurczak, Tetrahedron: Asymmetry, 2000, 11, 4581 CrossRef CAS.
- S. Yokoshima, H. Tokuyama and T. Fukuyama, Angew. Chem., Int. Ed., 2000, 39, 4073 CrossRef CAS.
- A. S. Raw and E. B. Jang, Tetrahedron, 2000, 56, 3285 CrossRef CAS.
- M. C. Carreño, J. L. G. Ruano, C. Z. Remor and A. Urbano, Tetrahedron: Asymmetry, 2000, 11, 4279 CrossRef CAS.
- M. C. Carreño, J. L. G. Ruano, A. Urbano, C. Z. Remor and Y. Arroyo, J. Org. Chem., 2000, 65, 453 CrossRef CAS.
- J. L. G. Ruano, C. Alemparte, A. M. M. Castro, H. Adams and J. H. R. Ramos, J. Org. Chem., 2000, 65, 7938 CrossRef.
- M. C. Carreño, S. García-Cerrada, A. Urbano and C. D. Vitta, J. Org. Chem., 2000, 65, 4355 CrossRef CAS.
- J. L. G. Ruano, F. Bercial, A. Fraile, A. M. M. Castro and M. R. Martín, Tetrahedron: Asymmetry, 2000, 11, 4737 CrossRef.
- M. A. Brimble, R. J. Elliott and J. F. McEwan, Aust. J. Chem., 2000, 53, 571 CrossRef CAS.
- G. B. Jones and M. Guzel, Tetrahedron Lett., 2000, 41, 4695 CrossRef CAS.
- E. J. Enholm and S. Jiang, J. Org. Chem., 2000, 65, 4756 CrossRef CAS.
- Y. Huang, P. E. Sonnet, P. J. Carroll and D. R. Dalton, Tetrahedron Lett., 2000, 41, 2519 Search PubMed.
- M. J. Burke, M. M. Allan, M. Parvez and B. A. Keay, Tetrahedron: Asymmetry, 2000, 11, 2733 CrossRef CAS.
- R. Chinchilla, L. R. Falvello, N. Galindo and C. Nájera, J. Org. Chem., 2000, 65, 3034 CrossRef CAS.
- T. Abellán, C. Nájera and J. M. Sansano, Tetrahedron: Asymmetry, 2000, 11, 1051 CrossRef CAS.
- M. T. Aranda, M. C. Aversa, A. Barattucci, P. Bonaccorsi, M. C. Carreño, M. B. Cid and J. L. G. Ruano, Tetrahedron: Asymmetry, 2000, 11, 1217 CrossRef CAS.
- J. M. Janey, T. Iwama, S. A. Kozmin and V. H. Rawal, J. Org. Chem., 2000, 65, 9059 CrossRef CAS.
- S. Pornpakakul, R. G. Pritchard and R. J. Stoodley, Tetrahedron Lett., 2000, 41, 2691 CrossRef CAS.
- N. S. Trotter, D. S. Larsen, R. J. Stoodley and S. Brooker, Tetrahedron Lett., 2000, 41, 8957 CrossRef CAS.
- R. Pedrosa, C. Andrés and J. Nieto, J. Org. Chem., 2000, 65, 831 CrossRef CAS.
- J. C. J. Benningshof, R. H. Blaauw, A. E. van Ginkel, F. P. J. T. Rutjes, J. Fraanje, K. Goubitz, H. Schenk and H. Hiemstra, Chem. Commun., 2000, 1465 RSC.
- J. Jauch, Angew. Chem., Int. Ed., 2000, 39, 2764 CrossRef CAS.
- P. D. Bailey, I. M. McDonald, G. M. Rosair and D. Taylor, Chem. Commun., 2000, 2451 RSC.
- C. Hedberg, P. Pinho, P. Roth and P. G. Andersson, J. Org. Chem., 2000, 65, 2810 CrossRef CAS.
- R. Paugam, E. Valenciennes, L. L. Coz-Bardol, J.-C. Garde, L. Wartski, M. Lance and M. Nierlich, Tetrahedron: Asymmetry, 2000, 11, 2509 CrossRef CAS.
- R. W. Ware Jr. and S. B. King, J. Org. Chem., 2000, 65, 8725 CrossRef CAS.
- A. Hall, P. D. Bailey, D. C. Rees, G. M. Rosair and R. H. Wightman, J. Chem. Soc., Perkin Trans. 1, 2000, 329 RSC.
- A. Defoin, M. Joubert, J.-M. Heuchel, C. Strehler and J. Streith, Synthesis, 2000, 1719 CrossRef CAS.
- M. Joubert, A. Defoin, C. Tarnus and J. Streith, Synlett, 2000, 1366 CAS.
- I. Cabanal-Duvillard, J.-F. Berrien, L. Ghosez, H.-P. Husson and J. Royer, Tetrahedron, 2000, 56, 3763 CrossRef CAS.
- I. Cabanal-Duvillard, J.-F. Berrien and J. Royer, Tetrahedron: Asymmetry, 2000, 11, 2525 CrossRef CAS.
- R. Fernández, A. Ferrete, J. M. Lassaletta, J. M. Llera and A. Monge, Angew. Chem., Int. Ed., 2000, 39, 2893 CrossRef CAS.
- R. Saul, J. Kopf and P. Köll, Tetrahedron: Asymmetry, 2000, 11, 423 CrossRef CAS.
- S. N. Joshi, A. R. A. S. Deshmukh and B. M. Bhawal, Tetrahedron: Asymmetry, 2000, 11, 1477 CrossRef CAS.
- I. Ojima, S. Lin, T. Inoue, M. L. Miller, C. P. Borella, X. Geng and J. J. Walsh, J. Am. Chem. Soc., 2000, 122, 5343 CrossRef CAS.
- K. Karupaiyan, V. G. Puranik, A. R. A. S. Desmukh and B. M. Bhawal, Tetrahedron, 2000, 56, 8555 CrossRef CAS.
- E. Falb, A. Nudelman, H. E. Gottlieb and A. Hassner, Eur. J. Org. Chem., 2000, 645 CrossRef CAS.
- P. J. Zimmermann, I. Blanarikova and V. Jäger, Angew. Chem., Int Ed., 2000, 39, 910 CrossRef CAS.
- H. Yamamoto, S. Watanabe, K. Kadotani, M. Hasegawa, M. Noguchi and S. Kanemasa, Tetrahedron Lett., 2000, 41, 3131 CrossRef CAS.
- G. Faita, A. Paio, P. Quadrelli, F. Rancati and P. Seneci, Tetrahedron Lett., 2000, 41, 1265 CrossRef CAS.
- K.-S. Yang, J.-C. Lain, C.-H. Lin and K. Chen, Tetrahedron Lett., 2000, 41, 1453 CrossRef CAS.
- M. Mauduit, C. Kouklovsky and Y. Langlois, Eur. J. Org. Chem., 2000, 1595 CrossRef CAS.
- U. Chiacchio, A. Rescifina, A. Corsaro, V. Pistarà, G. Romeo and R. Romeo, Tetrahedron: Asymmetry, 2000, 11, 2045 CrossRef CAS.
- O. Tamura, K. Gotanda, J. Yoshino, Y. Morita, R. Terashima, M. Kikuchi, T. Miyawaki, N. Mita, M. Yamashita, H. Ishibashi and M. Sakamoto, J. Org. Chem., 2000, 65, 8544 CrossRef CAS.
- P. Merino, S. Anoro, S. Franco, F. L. Merchan, T. Tejero and V. Tuñon, J. Org. Chem., 2000, 65, 1590 CrossRef CAS.
- M. C. Bagley and W. Oppolzer, Tetrahedron: Asymmetry, 2000, 11, 2625 CrossRef CAS.
- W. Oppolzer and C. G. Bochet, Tetrahedron: Asymmetry, 2000, 11, 4761 CrossRef CAS.
- S. Cicchi, P. Ponzuoli, A. Goti and A. Brandi, Tetrahedron Lett., 2000, 41, 1583 CrossRef CAS.
- F. M. Cordero, C. Faggi, F. De Sarlo and A. Brandi, Eur. J. Org. Chem., 2000, 3595 CrossRef CAS.
- S. E. Denmark and A. R. Hurd, J. Org. Chem., 2000, 65, 2875 CrossRef CAS.
- S. E. Denmark and B. Herbert, J. Org. Chem., 2000, 65, 2887 CrossRef CAS.
- M. G. B. Drew, L. M. Harwood, D. W. Price, M.-S. Choi and G. Park, Tetrahedron Lett., 2000, 41, 5077 CrossRef CAS.
- O. Tamura, S. Yoshida, H. Sugita, N. Mita, Y. Uyama, N. Morita, M. Ishiguro, T. Kawasaki, H. Ishibashi and M. Sakamoto, Synlett, 2000, 1553 CAS.
- P. R. Sebahar and R. M. Williams, J. Am. Chem. Soc., 2000, 122, 5666 CrossRef CAS.
- F. Roussi, A. Chauveau, M. Bonin, L. Micouin and H.-P. Husson, Synthesis, 2000, 1170 CrossRef CAS.
- G. A. Whitlock and E. M. Carreira, Helv. Chim. Acta, 2000, 83, 2007 CrossRef CAS.
- H. Sasaki and E. M. Carreira, Synthesis, 2000, 135 CrossRef CAS.
- B. Brown and L. S. Hegedus, J. Org. Chem., 2000, 65, 1865 CrossRef CAS.
- P. Del Buttero, C. Baldoli, G. Molteni and T. Pilati, Tetrahedron: Asymmetry, 2000, 11, 1927 CrossRef CAS.
- M. Pourashraf, P. Delair, M. O. Rasmussen and A. E. Greene, J. Org. Chem., 2000, 65, 6966 CrossRef CAS.
- C. Palomo, J. M. Aizpurua, R. Galarza, A. Benito, U. K. Khamrai, U. Eikeseth and A. Linden, Tetrahedron, 2000, 56, 5563 CrossRef CAS.
- K. Hiroi and A. Yamada, Tetrahedron: Asymmetry, 2000, 11, 1835 CrossRef CAS.
- O. Meth-Cohn, D. J. Williams and Y. Chen, Chem. Commun., 2000, 495 RSC.
- J. Raczko, M. Achmatowicz, P. Kwiatkowski, C. Chapuis, Z. Urbańczyk-Lipkowska and J. Jurczak, Tetrahedron: Asymmetry, 2000, 11, 1027 CrossRef CAS.
- Y. Takemoto, Y. Baba, G. Saha, S. Nakao, C. Iwata, T. Tanaka and T. Ibuka, Tetrahedron Lett., 2000, 41, 3653 CrossRef CAS.
- W. Adam, K. Peters, E.-M. Peters and S. B Schambony, J. Am. Chem. Soc., 2000, 122, 7610 CrossRef CAS.
- K. C. Nicolaou, J. Li and G. Zenke, Helv. Chim. Acta, 2000, 83, 1977 CrossRef CAS.
- B. R. Aavula, Q. Cui and E. A. Mash, Tetrahedron: Asymmetry, 2000, 11, 4681 CrossRef CAS.
- A. Mamai and J. S. Madalengoitia, Tetrahedron Lett., 2000, 41, 9009 CrossRef CAS.
- J. E. A. Luithle and J. Pietruszka, Eur. J. Org. Chem., 2000, 2557 CrossRef CAS.
- J. Pietruszka and A. Witt, J. Chem. Soc., Perkin Trans. 1, 2000, 4293 RSC.
- Q. Wang, M. F. Mayer, C. Brennan, F. Yang, M. M. Hossain, D. S. Grubisha and D. Bennett, Tetrahedron, 2000, 56, 4881 CrossRef CAS.
- A. B. Charette, J.-F. Marcoux, C. Molinaro, A. Beauchemin, C. Brochu and E. Isabel, J. Am. Chem. Soc., 2000, 122, 4508 CrossRef CAS.
- D. K. Heimbach, R. Fröhlich, B. Wibbeling and D. Hoppe, Synlett, 2000, 950 CAS.
- M. K. McKay and J. R. Green, Can. J. Chem., 2000, 78, 1629 CrossRef CAS.
- K. Kamikawa, T. Wantanabe, A. Daimon and M. Uemura, Tetrahedron, 2000, 56, 2325 CrossRef CAS.
- T. Watanabe, M. Shakadou and M. Uemura, Synlett, 2000, 1141 CAS.
- L. G. Monovich, Y. Le Huérou, M. Rönn and G. A. Molander, J. Am. Chem. Soc., 2000, 122, 52 CrossRef CAS.
- D. Enders, R. Peters, R. Lochtman and J. Runsink, Eur. J. Org. Chem., 2000, 2839 CrossRef CAS.
- R. Kitzler, L. Xiao and W. Weissensteiner, Tetrahedron: Asymmetry, 2000, 11, 3459 CrossRef CAS.
- D. Enders, R. Peters, R. Lochtman, G. Raabe, J. Runsink and J. W. Bats, Eur. J. Org. Chem., 2000, 3399 CrossRef CAS.
- G. Argouarch, O. Samuel, O. Riant, J.-C. Daran and H. B. Kagan, Eur. J. Org. Chem., 2000, 2893 CrossRef CAS.
- T. R. Burke Jr., D.-G. Liu and Y. Gao, J. Org. Chem., 2000, 65, 6288 CrossRef CAS.
- A. J. Pearson and P. O. Belmont, Tetrahedron Lett., 2000, 41, 1671 CrossRef CAS.
- M. C. Evans and R. L Johnson, Tetrahedron, 2000, 56, 9801 CrossRef CAS.
- S. Derrer, J. E. Davies and A. B. Holmes, J. Chem. Soc., Perkin Trans. 1, 2000, 2957 RSC.
- S. Derrer, J. E. Davies and A. B. Holmes, J. Chem. Soc., Perkin Trans. 1, 2000, 2943 RSC.
- P. C. B. Page, M. J. McKenzie, S. M. Allin and D. R. Buckle, Tetrahedron, 2000, 56, 9683 CrossRef CAS.
- C. C. Kotoris, W. Wen, A. Lough and S. D. Taylor, J. Chem. Soc., Perkin Trans. 1, 2000, 1271 RSC.
- F. A. Davis, H. Qi and G. Sundarababu, Tetrahedron, 2000, 56, 5303 CrossRef CAS.
- M. S. Idris, D. S. Larsen, R. G. Pritchard, A. Schofield, R. J. Stoodley and P. D. Tiffin, J. Chem. Soc., Perkin Trans. 1, 2000, 2195 RSC.
- S. A. Boyes and A. T. Hewson, J. Chem. Soc., Perkin Trans. 1, 2000, 2759 RSC.
- H. Fujioka, Y. Nagatomi, N. Kotoku, H. Kitagawa and Y. Kita, Tetrahedron, 2000, 56, 10141 CrossRef CAS.
- H. Fujioka, N. Kotoku, Y. Nagatomi and Y. Kita, Tetrahedron Lett., 2000, 41, 1829 CrossRef CAS.
- A. P. Esteves, A. M. Freitas, C. M. Raynor and R. J. Stoodley, J. Chem. Soc., Perkin Trans. 1, 2000, 1753 RSC.
- S. M. Allin, C. J. Northfield, M. I. Page and A. M. Z. Slawin, J. Chem. Soc., Perkin Trans. 1, 2000, 1715 RSC.
- A. Giardiná, T. Mecozzi and M. Petrini, J. Org. Chem., 2000, 65, 8277 CrossRef CAS.
- C. R. A. Godfrey, N. S. Simpkins and M. D. Walker, Synlett, 2000, 388 CAS.
- M. Cellier, Y. Gelas-Mialhe, H.-P. Husson, B. Perrin and R. Remuson, Tetrahedron: Asymmetry, 2000, 11, 3913 CrossRef CAS.
- M. D. Groaning and A. I. Meyers, Chem. Commun., 2000, 1027 RSC.
- C. Agami, S. Comesse and C. Kadouri-Puchot, J. Org. Chem., 2000, 65, 4435 CrossRef CAS.
- P. H. Dussault, T. K. Trullinger and S. Cho-Shultz, Tetrahedron, 56, 9213 Search PubMed.
- N. Maezaki, Y.-X. Li, K. Ohkubo, S. Goda, C. Iwata and T. Tanaka, Tetrahedron, 2000, 56, 4405 CrossRef CAS.
- N. Maezaki, A. Sakamoto, N. Nagahashi, M. Soejima, Y.-X. Li, T. Imamura, N. Kojima, H. Ohishi, K. Sakaguchi, C. Iwata and T. Tanaka, J. Org. Chem., 2000, 65, 3284 CrossRef CAS.
- J. Blanchet, M. Bonin, L. Micouin and H.-P. Husson, J. Org. Chem., 2000, 65, 6423 CrossRef CAS.
- J. M. Andrés, I. Herráiz-Sierra, R. Pedrosa and A. Pérez-Encabo, Eur. J. Org. Chem., 2000, 1719 CrossRef CAS.
- C. Spino, C. Beaulieu and J. Lafrenière, J. Org. Chem., 2000, 65, 7091 CrossRef CAS.
- C. Spino and C. Beaulieu, Angew. Chem., Int. Ed., 2000, 39, 1930 CrossRef CAS.
- C. Agami, F. Couty, G. Evano and H. Mathieu, Tetrahedron, 2000, 56, 367 CrossRef CAS.
- R. P. Singh, B. Twamley, L. Fabry-Asztalos, D. S. Matteson and J. M. Shreeve, J. Org. Chem., 2000, 65, 8123 CrossRef CAS.
- J. C. de la Rosa, N. Díaz and J. C. Carretero, Tetrahedron Lett., 2000, 41, 4107 CrossRef CAS.
- B. Guilloteau-Bertin, D. Compère, L. Gil, C. Marazano and B. C. Das, Eur. J. Org. Chem., 2000, 1391 CrossRef CAS.
- G. S. Currie, M. G. B. Drew, L. M. Harwood, D. J. Hughes, R. W. A. Luke and R. J. Vickers, J. Chem. Soc., Perkin Trans. 1, 2000, 2982 RSC.
- S. Madan, A. K. Sharma and S. S. Bari, Tetrahedron: Asymmetry, 2000, 11, 2267 CrossRef CAS.
- F. Jeanjean, G. Fournet, D. Le Bas and J. Goré, Eur. J. Org. Chem., 2000, 1297 CrossRef CAS.
- A. Nishida, F. Shirato and M. Nakagawa, Tetrahedron: Asymmetry, 2000, 41, 3789 CrossRef.
- F. Bois, D. Gardette and J.-C. Gramain, Tetrahedron Lett., 2000, 41, 8769 CrossRef CAS.
- G. Pandey, P. Das and P. Y. Reddy, Eur. J. Org. Chem., 2000, 657 CrossRef CAS.
- R. Pedrosa, C. Andrés, J. P. Duque-Soladana and C. D. Rosón, Tetrahedron: Asymmetry, 2000, 11, 2809 CrossRef CAS.
- R. Pedrosa, C. Andrés, J. P. Duque-Soladana and P. Mendiguchía, Eur. J. Org. Chem., 2000, 3727 CrossRef CAS.
- G. Chelucci, A. Saba, R. Valenti and A. Bacchi, Tetrahedron: Asymmetry, 2000, 11, 3449 CrossRef CAS.
- S. He, S. A. Kozmin and V. H. Rawal, J. Am. Chem. Soc., 2000, 122, 190 CrossRef CAS.
- S. Chandrasekhar and M. Sridhar, Tetrahedron: Asymmetry, 2000, 11, 3467 CrossRef CAS.
- J. Blanchet, M. Bonin, L. Micouin and H.-P. Husson, Tetrahedron Lett., 2000, 41, 8279 CrossRef CAS.
- A. Guarna, E. G. Occhiato, M. Pizzetti, D. Scarpi, S. Sisi and M. van Sterkenburg, Tetrahedron: Asymmetry, 2000, 11, 4227 CrossRef CAS.
- C. Agami, F. Couty and G. Evano, Tetrahedron Lett., 2000, 41, 8301 CrossRef CAS.
- T. Tsunoda, T. Nishii, M. Yoshizuka, C. Yamasaki, T. Suzuki and S. Itô, Tetrahedron Lett., 2000, 41, 7667 CrossRef CAS.
- A. Otaka, E. Mitsuyama, T. Kinoshita, H. Tamamura and N. Fujii, J. Org. Chem., 2000, 65, 4888 CrossRef CAS.
- M. Besson, P. Gallezot, S. Neto and C. Pinel, Tetrahedron: Asymmetry, 2000, 11, 1809 CrossRef CAS.
- T. J. Donohoe, A. A. Calabrese, C. A. Stevenson and T. Ladduwahetty, J. Chem. Soc., Perkin Trans. 1, 2000, 3724 RSC.
- T. M. T. Tuyet, T. Harada, K. Hashimoto, M. Hatsuda and A. Oku, J. Org. Chem., 2000, 65, 1335 CrossRef CAS.
- V. D. Rao and M. Periasamy, Tetrahedron: Asymmetry, 2000, 11, 1151 CrossRef CAS.
- R. W. Baker, R. V. Kyasnoor, M. V. Sargent, B. W. Skelton and A. H. White, Aust. J. Chem., 2000, 53, 487 CrossRef CAS.
- V. Narkevitch, K. Schenk and P. Vogel, Angew. Chem., Int. Ed., 2000, 39, 1806 CrossRef CAS.
- T. Tanaka, T. Azuma, X. Fang, S. Uchida, C. Iwata, T. Ishida, Y. In and N. Maezaki, Synlett, 2000, 33 CAS.
- M. Tiecco, L. Testaferri, C. Santi, C. Tomassini, F. Marini, L. Bagnoli and A. Temperini, Tetrahedron: Asymmetry, 2000, 11, 4645 CrossRef CAS.
- M. Calmès, F. Escale, C. Glot, M. Rolland and J. Martinez, Eur. J. Org. Chem., 2000, 2459 CrossRef CAS.
- B. M. Trost and G. A. Doherty, J. Am. Chem. Soc., 2000, 122, 3801 CrossRef CAS.
- P. Hall, J. Brun, D. Denni and R. Metternich, Synlett, 2000, 315 CAS.
- M. Banwell, A. Edwards, J. Harvey, D. Hockless and A. Willis, J. Chem. Soc., Perkin Trans. 1, 2000, 2175 RSC.
- H. Kim and H. M. R. Hoffmann, Eur. J. Org. Chem., 2000, 2195 CrossRef CAS.
- J. Busch-Petersen and E. J. Corey, Tetrahedron Lett., 2000, 41, 6941 CrossRef.
- K. W. Henderson, W. J. Kerr and J. H. Moir, Chem. Commun., 2000, 479 RSC.
- A. M. Montaña, D. Fernández, R. Pagès, A. C. Filippou and G. Kociok-Köhn, Tetrahedron, 2000, 56, 425 CrossRef CAS.
- K. Takasu, K. Misawa, M. Yamada, Y. Furuta, T. Taniguchi and M. Ihara, Chem. Commun., 2000, 1739 RSC.
- M. Majewski and P. Nowak, J. Org. Chem., 2000, 65, 5152 CrossRef CAS.
- M. Majewski, M. DeCaire, P. Nowak and F. Wang, Synlett, 2000, 1321 CAS.
- A. Kee, P. O'Brien, C. D. Pilgram and S. T. Watson, Chem. Commun., 2000, 1521 RSC.
- S. Barrett, P. O'Brien, H. C. Steffens, T. D. Towers and M. Voith, Tetrahedron, 2000, 56, 9633 CrossRef.
- S. Pache, C. Botuha, R. Franz, E. P. Kündig and J. Einhorn, Helv. Chim. Acta, 2000, 83, 2436 CrossRef CAS.
- T. Hata, H. Koide and M. Uemura, Synlett, 2000, 1145 CAS.
- P. Beak, D. R. Anderson, M. D. Curtis, J. M. Laumer, D. J. Pippel and G. A. Weisenburger, Acc. Chem. Res., 2000, 33, 715 CrossRef CAS.
- A. Deiters, R. Fröhlich and D. Hoppe, Angew. Chem., Int. Ed., 2000, 39, 2105 CrossRef CAS.
- K. R. K. Prasad and D. Hoppe, Synlett, 2000, 1067 CAS.
- K. B. Wiberg and W. F. Bailey, Angew. Chem., Int. Ed., 2000, 39, 2127 CrossRef CAS.
- W. F. Bailey and M. J. Mealy, J. Am. Chem. Soc., 2000, 122, 6787 CrossRef CAS.
- G. S. Gil and U. M. Groth, J. Am. Chem. Soc., 2000, 122, 6789 CrossRef.
- S. Nakamura, R. Nakagawa, Y. Watanabe and T. Toru, Angew. Chem., Int. Ed., 2000, 39, 353 CrossRef CAS.
- K. Nagata, S. Matsukawa and T. Imamoto, J. Org. Chem., 2000, 65, 4185 CrossRef CAS.
- A. Alexakis, E. Vrancken and P. Mangeney, J. Chem. Soc., Perkin Trans. 1, 2000, 3354 RSC.
- S. Nakamura, R. Nakagawa, Y. Watanabe and T. Toru, J. Am. Chem. Soc., 2000, 122, 11340 CrossRef CAS.
- K. Tomooka, L.-F. Wang, F. Okazaki and T. Nakai, Tetrahedron Lett., 2000, 41, 6121 CrossRef CAS.
- M. Imai, A. Hagihara, H. Kawasaki, K. Manabe and K. Koga, Tetrahedron, 2000, 56, 179 CrossRef CAS.
- D. A. Evans, J. S. Johnson and E. J. Olhava, J. Am. Chem. Soc., 2000, 122, 1635 CrossRef CAS.
- D. Taniyama, M. Hasegawa and K. Tomioka, Tetrahedron Lett., 2000, 41, 5533 CrossRef CAS.
- M. Hasegawa, D. Taniyama and K. Tomioka, Tetrahedron, 2000, 56, 10153 CrossRef CAS.
- S. E. Denmark and C. M. Stiff, J. Org. Chem., 2000, 65, 5875 CrossRef CAS.
- F. Xu, R. A. Reamer, R. Tillyer, J. M. Cummins, E. J. J. Grabowski, P. J. Reider, D. B. Collum and J. C. Huffman, J. Am. Chem. Soc., 2000, 122, 11212 CrossRef CAS.
- M. M. Rogic, J. Org. Chem., 2000, 65, 6868 CrossRef CAS.
- K. C. Nicolaou, Y. Li, B. Weyershausen and H. Wei, Chem. Commun., 2000, 307 RSC.
- C. B. Lee, T.-C. Chou, X.-G. Zhang, Z.-G. Wang, S. D. Kuduk, M. D. Chappell, S. J. Stachel and S. J. Danishefsky, J. Org. Chem., 2000, 65, 6525 CrossRef CAS.
- D. R. Williams, G. S. Cortez, S. L. Bogen and C. M. Rojas, Angew. Chem., Int. Ed., 2000, 39, 4612 CrossRef CAS.
- C. Hertweck and W. Boland, J. Org. Chem., 2000, 65, 2458 CrossRef CAS.
- M. Eggen, C. J. Mossman, S. B. Buck, S. K. Nair, L. Bhat, S. M. Ali, E. A. Reiff, T. C. Boge and G. I. Georg, J. Org. Chem., 2000, 65, 7792 CrossRef CAS.
- K. C. Nicolaou, F. Murphy, S. Barluenga, T. Ohshima, H. Wei, J. Xu, D. L. F. Gray and O. Baudoin, J. Am. Chem. Soc., 2000, 122, 3830 CrossRef CAS.
- P. V. Ramachandran, M. V. R. Reddy and H. C. Brown, Tetrahedron Lett., 2000, 41, 583 CrossRef CAS.
- M. B. Andrus, T. M. Turner, Z. E. Sauna and S. V. Ambudkar, J. Org. Chem., 2000, 65, 4973 CrossRef CAS.
- S. Girard, R. J. Robins, J. Villiéras and J. Lebreton, Tetrahedron Lett., 2000, 41, 9245 CrossRef CAS.
- A. G. M. Barrett, J. C. Beall, D. C. Braddock, K. Flack, V. C. Gibson and M. M. Salter, J. Org. Chem., 2000, 65, 6508 CrossRef CAS.
- D. J. S. Kumar, S. Madhavan, P. V. Ramachandran and H. C. Brown, Tetrahedron: Asymmetry, 2000, 11, 4629 CrossRef CAS.
- W. R. Roush, A. N. Pinchuk and G. C. Micalizio, Tetrahedron Lett., 2000, 41, 9413 CrossRef CAS.
- A. G. M. Barrett, D. C. Braddock, P. D. de Koning, A. J. P. White and D. J. Williams, J. Org. Chem., 2000, 65, 375 CrossRef CAS.
- J. M. Chong, L. Shen and N. J. Taylor, J. Am. Chem. Soc., 2000, 122, 1822 CrossRef CAS.
- F. Baburi, V. Fiandanese, G. Marchese and A. Punzi, Tetrahedron, 2000, 56, 327 CrossRef.
- F.-E. Chen, Y.-D. Huang, H. Fu, Y. Cheng, D.-M. Zhang, Y.-Y. Li and Z.-Z. Peng, Synthesis, 2000, 2004 CrossRef CAS.
- U. Gran, O. Wennerström and G. Westman, Tetrahedron: Asymmetry, 2000, 11, 3027 CrossRef CAS.
- N. Kanomata and T. Nakata, J. Am. Chem. Soc., 2000, 122, 4563 CrossRef CAS.
- J. Li, Y.-C. Liu, J.-G. Deng, X.-Z. Li, X. Cui and Z. Li, Tetrahedron: Asymmetry, 2000, 11, 2677 CrossRef CAS.
- D. Murali, B. Singaram and H. C. Brown, Tetrahedron: Asymmetry, 2000, 11, 4831 CrossRef CAS.
- J. Cossy, C. Willis, V. Bellosta and S. Bouzbouz, Synlett, 2000, 1461 CAS.
- S. Bouzbouz and J. Cossy, Tetrahedron Lett., 2000, 41, 3363 CrossRef CAS.
- S. Bouzbouz, F. Pradaux, J. Cossy, C. Ferroud and A. Falguières, Tetrahedron Lett., 2000, 41, 8877 CrossRef CAS.
- Z. Wu, F. Zhang and S. J. Danishefsky, Angew. Chem., Int. Ed., 2000, 39, 4505 CrossRef CAS.
- J. A. Marshall, K. Gill and B. M. Seletsky, Angew. Chem., Int. Ed., 2000, 39, 953 CrossRef CAS.
- S. Kiyooka and K. A. Shahid, Tetrahedron Lett., 2000, 41, 2633 CrossRef CAS.
- P. Kocienski, R. Narquizian, P. Raubo, C. Smith, L. J. Farrugia, K. Muir and F. T. Boyle, J. Chem. Soc., Perkin Trans. 1, 2000, 2357 RSC.
- F. Yokokawa, J. Izumi, J. Omata and T. Shioiri, Tetrahedron, 2000, 56, 3027 CrossRef CAS.
- S. Kiooka, M. A. Hena, T. Yabukami, K. Murai and F. Goto, Tetrahedron Lett., 2000, 41, 7511 CrossRef.
- S. Kiyooka, K. Goh, Y. Nakamura, H. Takesue and M. A. Hena, Tetrahedron Lett., 2000, 41, 6599 CrossRef CAS.
- E. J. Corey and S. Choi, Tetrahedron Lett., 2000, 41, 2765 CrossRef CAS.
- E. J. Corey and S. Choi, Tetrahedron Lett., 2000, 41, 2769 Search PubMed.
- I. Paterson, G. F. Florence, K. Gerlach and J. P. Scott, Angew. Chem., Int. Ed., 2000, 39, 377 CrossRef.
- T. Itoh, H. Inoue and S. Emoto, Bull. Chem. Soc. Jpn., 2000, 73, 409 CrossRef CAS.
- M. Calmes, F. Escale, E. Juan, M. Rolland and J. Martinez, Tetrahedron: Asymmetry, 2000, 11, 1263 CrossRef CAS.
- M. Müller, K. Lamottke, E. Löw, E. Magar-Veenstra and W. Steglich, J. Chem. Soc., Perkin Trans. 1, 2000, 2483 RSC.
- T. Ooi, K. Takaya, T. Miura, H. Ichikawa and K. Maruoka, Synlett, 2000, 1133 CAS.
- G. Karig, A. Fuchs, A. Busing, T. Brandsletter, S. Scherer, J. W. Bats, A. Eschenmoser and G. Quinkert, Helv. Chim. Acta, 2000, 83, 1049 CrossRef CAS.
- S. M. Moharram, G. Hirai, K. Koyama, H. Oguri and M. Hirama, Tetrahedron Lett., 2000, 41, 6669 CrossRef CAS.
- A. Bayer and O. R. Gautun, Tetrahedron Lett., 2000, 41, 3743 CrossRef CAS.
- T. Bach, H. Bergmann and K. Harms, Angew. Chem., Int. Ed., 2000, 39, 2302 CrossRef CAS.
- C. D. Davies, S. P. Marsden and E. S. E. Stokes, Tetrahedron Lett., 2000, 41, 4229 CrossRef CAS.
- A. R. Kennedy, W. J. Kerr, D. M. Lindsay, J. S. Scott and S. P. Watson, J. Chem. Soc., Perkin Trans. 1, 2000, 4366 RSC.
- J. Castro, A. Moyano, M. A. Pericàs, A. Riera, A. Alvarez-Larena and J. F. Piniella, J. Am. Chem. Soc., 2000, 122, 7944 CrossRef CAS.
- D. R. Carbery, W. J. Kerr, D. M. Lindsay, J. S. Scott and S. P. Watson, Tetrahedron Lett., 2000, 41, 3235 CrossRef CAS.
- X. Verdaguer, A. Moyano, M. A. Pericàs, A. Piera, M. A. Maestro and J. Mahía, J. Am. Chem. Soc., 2000, 122, 10242 CrossRef CAS.
- A. J. Fletcher, D. T. Rutherford and S. D. R. Christie, Synlett, 2000, 1040 CAS.
- W. J. Kerr, D. M. Lindsay, E. M. Rankin, J. S. Scott and S. P. Watson, Tetrahedron Lett., 2000, 41, 3229 CrossRef CAS.
- I. Coldham and G. P. Vennall, Chem. Commun., 2000, 1569 RSC.
- R. W. Hoffmann, B. Hölzer, O. Knopff and K. Harms, Angew. Chem., Int. Ed., 2000, 39, 3072 CrossRef CAS.
- C. N. Farthing and S. P. Marsden, Tetrahedron Lett., 2000, 41, 4235 CrossRef CAS.
- P. E. Morgan and R. McCague and A Whiting, J. Chem. Soc., Perkin Trans. 1, 2000, 515 RSC.
- S. Saito, M. Nakadai and H. Yamamoto, Synlett, 2000, 1107 CAS.
- Y. Ukaji, Y. Yoshida and K. Inomata, Tetrahedron: Asymmetry, 2000, 11, 733 CrossRef CAS.
- A. de Meijere, B. Stecker, A. Kourdioukov and C. M. Williams, Synthesis, 2000, 929 CrossRef CAS.
- E. Vedejs, A. W. Kruger, N. Lee, S. T. Sakata, M. Stec and E. Suna, J. Am. Chem. Soc., 2000, 122, 4602 CrossRef CAS.
- S. Nakamura, M. Kaneeda, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2000, 122, 8120 CrossRef CAS.
- A. Cuenca, M. Medio-Simón, G. A. Aguilar, D. Weibel, A. K. Beck and D. Seebach, Helv. Chim. Acta, 2000, 83, 3153 CrossRef CAS.
- Y. Nakamura, S. Takeuchi, Y. Ohgo and D. P. Curran, Tetrahedron, 2000, 56, 351 CrossRef.
- A. J. Burton, J. P. Graham and N. S. Simpkins, Synlett, 2000, 1640 CAS.
- S. Matsukawa and T. Imamoto, J. Am. Chem. Soc., 2000, 122, 12659 CrossRef CAS.
- J. Eames and N. Weerasooriya, Tetrahedron Lett., 2000, 41, 521 CrossRef CAS.
- Z. Liu, N. Shibata and Y. Takeuchi, J. Org. Chem., 2000, 65, 7583 CrossRef CAS.
- L. E. Overman, J. F. Larrow, B. A. Stearns and J. M. Vance, Angew. Chem., Int. Ed., 2000, 39, 213 CrossRef CAS.
- C.-C. Wang, J. J. Li, H.-C. Huang, L. F. Lee and D. B. Reitz, J. Org. Chem., 2000, 65, 2711 CrossRef CAS.
- V. V. Zhdankin, J. T. Smart, P. Zhao and P. Kiprof, Tetrahedron Lett., 2000, 41, 5299 CrossRef CAS.
- S. Nakamura, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2000, 122, 8131 CrossRef CAS.
- K. Manabe and S. Kobayashi, Chem. Commun., 2000, 669 RSC.
- Y. Miyake, H. Takada, K. Ohe and S. Uemura, J. Chem. Soc., Perkin Trans. 1, 2000, 1595 RSC.
- Y. Lu and G. Just, Tetrahedron, 2000, 56, 4355 CrossRef CAS.
- M. Tiecco, L. Testaferri, C. Santi, C. Tomassini, F. Marini, L. Bagnoli and A. Temperini, Eur. J. Org. Chem., 2000, 3451 CrossRef CAS.
- K. Hiroi and M. Ishii, Tetrahedron Lett., 2000, 41, 7071 CrossRef CAS.
- D. Enders and E. C. Ullrich, Tetrahedron: Asymmetry, 2000, 11, 3861 CrossRef CAS.
- S. Cossu, O. De Lucchi, P. Peluso and R. Volpicelli, Tetrahedron Lett., 2000, 41, 7263 CrossRef CAS.
- I. Michieletto, F. Fabris and O. De Lucchi, Tetrahedron: Asymmetry, 2000, 11, 2835 CrossRef CAS.
- H. Watanabe, D. Itoh and T. Kitahara, Synthesis, 2000, 1925 CrossRef CAS.
- K. Ishihara, H. Nakamura and H. Yamamoto, Synlett, 2000, 1245 CAS.
- A. G. Al-Sehemi, R. S. Atkinson, J. Fawcett and D. R. Russell, Tetrahedron Lett., 2000, 41, 2239 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |