Photochemical transformations of 5-halogeno-4-thiouridines
Grażyna Wenskaa, Katarzyna Taras-Goślińskaa, Katarzyna Lamparska-Kupsika, Bohdan Skalski*a, Maria Gdanieca and Zofia Gdaniecb
aFaculty of Chemistry, A. Mickiewicz University, Grunwaldzka 6, 60-780 Poznań, Poland. E-mail: bskalski@amu.edu.pl; Fax: 48-61 8658008; Tel: 48-61 829351
bInstitute of Bioorganic Chemistry, Polish Academy of Sciences, Poznań, Poland
First published on 6th December 2001
Abstract
A series of 2′,3′,5′-tri-O-acetyl-5-halogeno-4-thiouridines, where halogen = Br, Cl, F, are synthesized and their photochemical transformations upon irradiation with near-UV light (λ > 300 nm) in aqueous acetonitrile solutions investigated. The main photochemical pathways in each case involve intermolecular additions leading to three main photoproducts with different relative distributions. The photoproducts are isolated and their structures determined based on MS, 1H and 13C NMR and UV spectral data.
Introduction
Because of its structural similarity to uracil and its high photoreactivity with nucleobases and amino acid residues, combined with the possibility of selective excitation (λmax ≈ 330 nm) within various nucleoprotein complexes, the 4-thiouracil (s4U) chromophore and its derivatives have attracted considerable interest in recent years as efficient photocrosslinking agents in the studies of nucleic acids structure1 and their interactions with proteins.2,3 5-Halogenouracils, and especially 5-bromo and 5-iodo derivatives, belong to a second group of photochemically active pyrimidines most frequently used in this area of research.2 Unlike s4U, both halogenouridines absorb only weakly above 300 nm; however, they photoreact efficiently with various amino acid derivatives. In a search for new photolabels we turned our attention to 5-halogeno-4-thiouracil chromophores bearing both of the above mentioned photochemically active functionalities. Some of these novel uracil derivatives have been synthesized recently in a search for new, more effective antitumor agents as well as simple models for the mechanistic studies of some enzymatic reactions. They include the bases 5-bromo-, 5-chloro- and 5-fluoro-4-thiouracil4,5 and nucleosides 1-(α-D-arabinofuranosyl)-5-chloro-4-thiouracil,6 2′-deoxy-5-fluoro-4-thiouridine and 5-fluoro-4-thiouridine 5′-monophosphate.7As a part of our research goal to evaluate the potential of the 5-halogeno-4-thiouracil chromophores as photocrosslinking agents in nucleic acids we have undertaken the synthesis and detailed photochemical study of several 2′,3′,5′-tri-O-acetyl-5-halogeno-4-thiouridines 1a–c. In this paper we report on the isolation and characterization of the photoproducts resulting from irradiation of 1a–c in acetonitrile–water solutions.
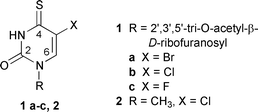
Results and discussion
2′,3′,5′-Tri-O-acetyl-5-halogeno-4-thiouridines 1a–c were synthesized from their 4-oxo analogues by reaction with P2S5 according to general thionation procedure.8 The structures of these novel thiouridine derivatives were confirmed by spectroscopic methods (Experimental section). Solutions of 1a–c (5 × 10−4 M) in a 50 ∶ 50 (v/v) acetonitrile–water mixture were irradiated at λ > 300 nm under anaerobic conditions. The photoreactions were monitored by the disappearance of the thiocarbonyl absorption band in the UV spectra of 1a–c (λmax > 330 nm, see Fig. 1) and by HPLC. The rates and the quantum yields of disappearance of 1a–c were found to depend on the nature of the halogen substituent and decrease in the order of 1c (ϕ = 3.4 ± 0.1 × 10−3) > 1a (ϕ = 1.8 ± 0.1 × 10−3) > 1b (ϕ = 0.5 ± 0.1 × 10−3). For preparative purposes, the irradiations were continued until ca. 40–50% conversion of the substrates was reached, as detected by HPLC. | ||
| Fig. 1 Changes in the absorption spectra of a solution of 1b in a 50 ∶ 50 (v/v) acetonitrile–water mixture during irradiation (λ > 300 nm) in the absence of oxygen. | ||
A typical HPLC profile obtained for the irradiated solution of 1b, revealing formation of three main photoproducts, 3, 4 and 5b, is shown in Fig. 2(A) while Fig. 2(B) shows their UV spectra. The photoproducts were isolated by preparative reversed-phase HPLC, and their structures were determined based on the HR-LSIMS, 1H and 13C NMR and UV spectral data. The HPLC-determined yields of the photoproducts, calculated in respect of the amount of the substrates consumed, are included in Scheme 1. Photoproducts 3 and 4 were identical to the photoproducts from irradiation of the solutions of the remaining two nucleosides 1a and 1c, which indicated that their formation involves loss of the halogen substituents.
 | ||
| Scheme 1 The structures and HPLC-determined yields of the photoadducts isolated from irradiated solutions of 1a–c and 2. The numbering system of atoms in the structures refers to the 1H and 13C NMR signals assignment. | ||
 | ||
| Fig. 2 (A) HPLC analysis after irradiation of 1b. The column was Waters Nova-Pak C18 60 Å 4 μm (4.6 × 250 mm), eluted with acetonitrile–water containing 10 mM ammonium acetate, with a linear gradient of 25–80% of acetonitrile over a period of 15 min. (B) Quantitative UV spectra of 1b and the photoproducts 3, 4 and 5b. | ||
The molecular formula of 3, determined as C30H32N4O16S by HR-LSIMS, indicates that its formation involves photoaddition of two substrate molecules and loss of hydrogen sulfide and both halogen substituents. The 1H NMR spectrum of 3 shows two non-exchangeable singlets at δ 9.18 and 8.31 and two easily recognizable sets of signals of the sugar protons. Both the chemical shifts and the appearance of the sugar proton multiplets of 3 do not differ significantly from those of the substrates 1a–c, indicating that the sugar residues in the former remain intact. The olefinic signals at δ 9.18 and 8.31 were assigned to H(6) and H(6′), respectively, on the basis of their NOEs with the anomeric protons of the neighboring sugar residues and by comparison of their chemical shifts with those of the corresponding protons in structurally related C(4)–C(5′)-connected bipyrimidines.9 The 13C NMR spectrum of 3 consists of 28 lines, most of which are attributable to carbon atoms of the two sugar moieties. The absence of a resonance in the region of δC 190–185, characteristic for a thiocarbonyl group, supports the suggestion that the sulfur atom in 3 is involved in a sulfide linkage. To simplify interpretation of the NMR spectra of 3, obscured by the resonances of the sugar carbons, we have synthesized a methyl analogue 6 of this photoproduct by irradiation of 5-chloro-1-methyl-4-thiouracil 2 (Scheme 1). Four singlets at δ 9.27, 8.65, 3.58 and 3.49 in the 1H NMR spectrum of 6 were assigned to H(6), H(6′), 1(CH3) and 1′(CH3), respectively. The unequivocal assignment of all carbon signals followed from the 1H–13C HMQC and 1H–13C HMBC spectra and is presented in Table 1. The key NMR evidence for the C(5)–C(4′) linkage of the two uracil rings in 6 are strong cross peaks of both H(6) and H(6′) to C(5) observed in the HMBC spectrum of the photoproduct.
| Comp. | Carbon atom (see Scheme 1 for locants) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (2) | (2′) | (4) | (4′) | (5) | (5′) | (6) | (6′) | CH3 (1) | CH3 (1′) | |
| 2 | 148.19 | 185.53 | 115.73 | 141.37 | 36.19 | |||||
| 6 | 153.53 | 155.01 | 179.98 | 163.87 | 109.42 | 108.30 | 149.56 | 144.73 | 39.22 | 39.10 |
| 7 | 154.55 | 155.04 | 173.49 | 165.07 | 101.61 | 107.68 | 158.51 | 141.77 | 26.58 | 38.66 |
The structure of photoproduct 6 was definitively established by X-ray analysis (Fig. 3).†
 | ||
| Fig. 3 ORTEP drawing of one of the two symmetry-independent molecules of compound 6. | ||
The structure of photoproduct 4 and its N-methyl analogue 7 was deduced from the MS, NMR and UV spectral data. Comparison of the molecular formulae of 4 and 7, found from exact mass measurement of the quasimolecular ions (M + Na+) in the HR-ESIMS spectra, with those of 3 and 6 respectively, indicates that the former contain one oxygen atom more. The 1H NMR spectrum of 7 in DMSO-d6 is very simple and displays only 3 singlets, at δH 8.62, 3.47 and 2.50, assigned to H(6′), CH3(1′) and CH3(1), respectively. A careful analysis of the 1H and 13C NMR spectra of 7 using the HMBC and HMBQ techniques enabled the unequivocal identification of all carbon signals (Table 1) and supported the proposed structure. The significant downfield shift of the C(6) signal (Δδ 8.95 ppm) together with an upfield shift of the CH3(1) signal (Δδ 12.64 ppm) in the case of 7, compared with the corresponding carbon signals of 6 (δC 149.56 and 39.22, respectively) is consistent with the presence of the carbonyl group in the 6-position of the former. As expected, in the case of 4 the presence of this group results in both a downfield shift of the neighboring anomeric proton signal (≈0.4 ppm) and a steric hindrance to rotation about the glycosidic bond. The latter is manifested by the reversible, temperature-dependent changes in the appearance of this signal, which is broad at room temperature and becomes sharp at higher temperatures (spectra not shown). A similar downfield shift (≈1 ppm) of the sugar anomeric proton signal in uracil 3-β-D-ribofuranoside as compared with the 1-β-D-ribofuranoside,10 as well as the existence of two rotational isomers of 3-β-D-glucopyranosyl-6-methyluracils, was demonstrated previously by 1H NMR spectroscopy.11 We must stress that although the chemical shifts, multiplicity and integration of all signals present in the 1H NMR spectra of 4 and 7 in D2O are fully compatible with their proposed structures, in neither case could an expected exchangeable-proton signal in the NMR spectra recorded in DMSO-d6 be observed. In the structural formula of photoproduct 7 (Scheme 1), which represents one of its several possible tautomeric forms, this H atom is connected to N(3). An exchangeable NH-proton signal was identified in the NMR spectra of several structurally related barbituric acid derivatives; however, this signal was reported to be solvent- and sample-concentration-dependent, being very broad or even not observed in dilute samples.12 Unfortunately, in the case of 4 and 7, both the choice of the solvent and the sample concentrations used in the NMR experiments were limited by their low solubility (<10 mg/0.5 mL in DMSO-d6).
Evidence supporting the presence of an easily exchangeable, acidic proton in 4 and 7 is the observed pH dependency of their UV spectra. Both compounds exhibit identical UV absorption spectra with a maximum at 342 nm in neutral aqueous solutions, which shifts to 383 nm upon acidifying the solutions to pH 3.0 (see Fig. 4). The reversibility of the spectral changes as well as the pH range where they are observed (pH 3–7) suggest that they result from a prototropic acid–base equilibrium and that 4 and 7 are weak acids. The value of pKa = 5.1 determined for 7 from the spectrophotometric titration falls in the range typical for ionization constants of barbituric acid derivatives.13
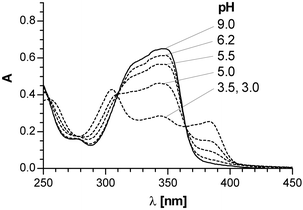 | ||
| Fig. 4 The pH dependence of the UV spectra of 7. | ||
Photoproduct 5a was found to have the molecular formula of C30H33BrN4O16S2, indicating that it results from an intermolecular coupling of two substrate molecules, accompanied by loss of an HBr molecule. The presence of two sugar residues in 5a is evident from the appearance of two sets of acetyl and ribosyl signals in the 1H and 13C NMR spectra. An exchangeable, broad signal at δ 9.58 and two sharp monoprotonic singlets at δ 7.99 and 7.98 in the low-field part of the 1H NMR spectrum, attributable to the N–H, H(6) and H(6′) pyrimidine protons, respectively, confirm the structure proposed for 5a. The appearance of only one carbon signal in the region δC 185–190 characteristic for the 4-thiouridine thiocarbonyl group,6,14 together with an intense long-wavelength absorption band at 329 nm in the UV spectrum of 5a, confirm the presence of the thiouracil residue in the molecule. Involvement of a second sulfur in a sulfide linkage between the two pyrimidine rings in 5a is supported by the presence of a resonance at δC 174.72 in the 13C NMR spectrum, attributable to C(4′)S. Photoproduct 5b, which was a minor constituent of the mixture obtained from irradiation of the chlorine-containing 4-thiouridine derivative 1b (Scheme 1), was identified by comparison of its UV and 1H NMR spectral data with those found for 5a and was further confirmed by HR-LSIMS spectroscopy (Experimental section). No formation of 5c, i.e. the fluorine-containing analogue of photoproducts 5a,b, was observed in the case of irradiation of 2′,3′,5′-tri-O-acetyl-5-fluoro-4-thiouridine 1c.
It is interesting to note that the adducts obtained by irradiation of 1a–c and 2 are structurally distinct from the bimolecular photoadduct, 5-(2′-oxo-1∶2-dihydropyrimidin-4′-yl)-4-thiouracil, formed by irradiation of 4-thiouracil in water,15 as well as similar (5)–(4′) and/or (6)–(4′) bipyrimidine products formed upon irradiation of s4U in the presence of pyrimidine bases.1 These adducts are believed to arise from [2 + 2] addition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond of s4U to the (5)–(6) CC bond of another pyrimidine, leading to thermally unstable thietane intermediates. It is reasonable to assume that a similar mechanism operates in the case of 5a,b,
whose formation involves coupling of two substrate molecules and loss of only one HX molecule, as well as in the case of photoproducts 3, 6, 4 and 7, which evidently result from a more complicated series of reactions involving loss of both halogen substituents and additional loss of the sulfur atom. Furthermore, the fact that, in each of the above photoadducts, the bridging sulfur atom has the same connectivity (C4–S–C5) indicates that they may arise from a common intermediate thietane. It should be noted, however, that under our experimental conditions we did not observe any unstable precursors of the photoproducts.
S double bond of s4U to the (5)–(6) CC bond of another pyrimidine, leading to thermally unstable thietane intermediates. It is reasonable to assume that a similar mechanism operates in the case of 5a,b,
whose formation involves coupling of two substrate molecules and loss of only one HX molecule, as well as in the case of photoproducts 3, 6, 4 and 7, which evidently result from a more complicated series of reactions involving loss of both halogen substituents and additional loss of the sulfur atom. Furthermore, the fact that, in each of the above photoadducts, the bridging sulfur atom has the same connectivity (C4–S–C5) indicates that they may arise from a common intermediate thietane. It should be noted, however, that under our experimental conditions we did not observe any unstable precursors of the photoproducts.
Experimental
5-Fluorouridine (Aldrich) was acetylated according to the reported, general procedure.16 2′,3′,5′-Tri-O-acetyl-5-chlorouridine,17 2′,3′,5′-tri-O-acetyl-5-bromouridine17 and 5-chloro-1-methyluracil17,18 were prepared according to the published procedures. TLC was performed using precoated plates (silica gel 60 PF254, 0.25 mm, Merck). Column chromatography was performed using Merck silica gel 60 (70–230 mesh). HPLC separations were performed on a Waters 600E instrument equipped with a Waters 991 Photodiode Array UV detector using Waters Nova-Pak C18 4 μm (4.6 × 250 mm) and XTerra RP18 3.5 μm (4.6 × 150 mm) columns at a flow rate of 0.8 mL min−1 for analytical runs and a Waters Nova Pack HRC18 RCM (25 × 100 mm) column at a flow rate of 3 mL min−1 for preparative separations. UV spectra were recorded on a Perkin-Elmer Lambda 17 spectrophotometer. The pH-dependent UV spectra were measured in sodium acetate–acetic acid (pH 3.4–6) and phosphate (pH 5.8–8) buffers. Extremes of pH made use of 0.1 M solutions of HCl and NaOH. 1H NMR, broadband-decoupled 13C and 1H, 13C 2D-HETCOR and NOE difference spectra were recorded on a Varian Unity 300 (300 MHz and 75 MHz respectively); HMBC, HMQC and temperature-dependent 1H NMR spectra on a Bruker AVANCE 600 (600 MHz and 150 MHz, respectively). All chemical shifts (δ) are in ppm relative to tetramethysilane as internal standard and coupling constants (J) are in Hz. High-resolution liquid matrix secondary-ion mass spectra (HR-LSIMS) were recorded in positive-ion mode on an AMD Intectra Model 604, and high-resolution electrospray ionization time-of-flight mass spectra (ESI-TOF-MS) on a Mariner PerSeptive Biosystems instrument.2′,3′,5′-Tri-O-acetyl-5-bromo-4-thiouridine 1a
2′,3′,5′-Tri-O-acetyl-5-bromouridine (0.898 g, 2 mmol) was dissolved in peroxide-free 1,4-dioxane (12 mL) and P2S5 (1.334 g, 6 mmol) was added. The mixture was refluxed until TLC analysis (CHCl3–CH3OH, 97 ∶ 3, v/v) showed complete disappearance of the substrate (4–5 h). Solvent was removed under reduced pressure and the residue was treated several times with CHCl3. The combined chloroform extracts were evaporated and the residue separated on silica gel. The column was eluted with CHCl3, followed by MeOH in CHCl3 [0–30% (v/v)]. Homogeneous fractions were collected and solvent was removed under reduced pressure to give 1a as a foam (0.688 g, 74%), λmax (H2O)/nm 339 (ε/dm3 mol−1 cm−1 16 700); δH (CDCl3) 10.06 (1 H, s, N–H exchangeable in D2O), 7.89 (1 H, s, 6-H), 6.05 (1 H, d, J 4.40, 1′-H), 5.40–5.32 (2 H, m, 2′- and 3′-H), 4.43–4.33 (3 H, m, 4′-, 5′- and 5″-H), 2.14 (3 H, s, 3, COMe), 2.13 (3 H, s, COMe), 2.12 (3 H, s, COMe); δC (CDCl3) 185.24 (4-C), 169.82, 169.35 and 169.32 (COMe), 146.79 (2-C), 134.11 (6-C), 108.32 (5-C), 87.60 (1′-C), 80.43 (2′-C), 73.17 (3′-C), 69.96 (4′-C), 62.78 (5′-C), 20.98, 20.53, 20.45 (COCH3); HR-LSIMS (m-nitrobenzyl alcohol) m/z 464.99649 [M(79Br) + H+], C15H1879BrN2O8S requires m/z 464.99673; m/z 466.99514 [M(81Br) + H+], C15H1881BrN2O8S requires m/z, 466.99469.2′,3′,5′-Tri-O-acetyl-5-chloro-4-thiouridine 1b and 2′,3′,5′-tri-O-acetyl-5-fluoro-4-thiouridine 1c
2′,3′,5′-Tri-O-acetyl-5-chlorouridine (0.809 g, 2 mmol) and 2′,3′,5′-tri-O-acetyl-5-fluorouridine (0.777 g, 2 mmol) were treated with P2S5 using the procedure described above for the synthesis of 1a. The reaction mixtures were worked up as above to give 1b (0.614 g, 73%) and 1c (0.615 g, 76%) as foams.1bλmax (H2O)/nm 339 (ε/dm3 mol−1 cm−1 16 800); δH (CDCl3) 10.13 (1 H, s, N–H exchangeable in D2O), 7.81 (1 H, s, 6-H), 6.05 (1 H, d, J 4.67, 1′-H), 5.39–5.31 (2 H, m, 2′- and 3′-H), 4.43–4.34 (3 H, m, 4′-, 5′- and 5″-H), 2.15 (3 H, s, 3, COMe), 2.14 (3 H, s, 3, COMe), 2.13 (3 H, s, 3, COMe); δC (CDCl3) 184.70 (4-C), 170.09, 169.63 and 169.59 (COMe), 146.77 (2-C), 131.61 (6-C), 119.27 (5-C), 87.15 (1′-C), 80.39 (2′-C), 73.16 (3′-C), 69.87 (4′-C), 62.71 (5′-C), 20.74, 20.38, 20.31(COCH3); HR LSIMS (m-nitrobenzyl alcohol) m/z 421.05044 (M + H+), C15H18ClN2O8S requires m/z 421.04724.
1cλmax (H2O)/nm 331 (ε/dm3 mol−1 cm−1 19 200); δH (CDCl3) 10.08 (1 H, s, N–H exchangeable in D2O), 7.55 (1 H, d, 3JH,F 4.5, 6-H), 6.05 (1 H, dd, J 1.2, J 4.5, 1′-H), 5.38–5.30 (2 H, m, 2′- and 3′-H), 4.43–4.33 (3 H, m, 4′-, 5′- and 5″-H), 2.18 (3 H, s, COMe), 2.13 (3 H, s, COMe), 2.12 (3 H, s, COMe); δC(CDCl3) 179.86 (d, 3JC,F 31, 4-C), 169.78, 169.39 and 169.36 (COMe), 147.23 (d, 1JC,F 227.3, 5-C), 146.39 (2-C), 118.86 (d, 3JC,F 41.2, 6-C), 87.66 (1′-C), 80.22 (2′-C), 72.97 (3′-C), 69.82 (4′-C), 62.76 (5′-C), 20.78, 20.55, 20.48 (COCH3); HR-LSIMS (m-nitrobenzyl alcohol) m/z 405.07702 (M + H+), C15H18FN2O8S requires m/z 405.07678.
5-Chloro-1-methyl-4-thiouracil 2
5-Chloro-1-methyluracil (0.722 g, 4.5 mmol) was treated with P2S5 analogously as described above for the synthesis of 1a–c. After 2.5 h of heating the reaction mixture was filtered and the filtrate evaporated to dryness. The residue was dissolved in a small amount of formic acid, applied to a silica gel column, and eluted with CHCl3 followed by CHCl3–CH3OH 10 ∶ 1 (v/v). Homogeneous fractions of product were collected, evaporated to dryness and the residue was finally recrystallized from hot water to give yellow crystals of title compound 2 (0.556 g, 70%), λmax (H2O)/nm 339 (ε/dm3 mol−1 cm−1 16 300); δH (DMSO-d6) 13.07 (1 H, br s, NH), 8.26 (6-H), 3.34 (3 H, s, NCH3); δC (DMSO-d6) 185.53 (4-C), 148.19 (2-C), 141.37 (6-C), 115.73 (5-C), 36.19 (CH3); HR-LSIMS (m-nitrobenzyl alcohol) m/z 176.98794 (M + H+), C5H5ClN2OS requires m/z 176.98826.Analytical-scale irradiations and quantum-yield determination
Solutions (2 mL) of 1a–c (5 × 10−4 M) in a 50 ∶ 50 (v/v) acetonitrile–water mixture were placed in a 1 × 1 cm UV cell, deoxygenated by bubbling argon, and irradiated on an optical bench using a 200 W high-pressure mercury lamp equipped with a glass filter λ > 330 nm or, for quantum-yield determination, with a 313 nm interference filter. The progress of the reaction was monitored by UV spectroscopy and HPLC. For the quantum-yield determination the samples were irradiated to low conversion (≈10%) of the substrates. The amount of 1a–c that reacted was determined by HPLC analysis. Hexan-2-one actinometry was used.19Preparative irradiations
1.5 mmol solutions of tri-O-acetyl-5-halogeno-4-thiouridines or 5-chloro-1-methyl-4-thiouracil in 50% aq. acetonitrile were irradiated in portions, in a 80 mL photoreactor with a 150W immersed high-pressure mercury lamp through a Pyrex filter under inert, argon atmosphere. The irradiations were continued until ≈40–50% of the substrates reacted as checked by HPLC. Irradiated solutions were collected and evaporated to dryness. The photoproducts were isolated from the residues either by preparative HPLC (5a,b) or by successive column chromatography on silica gel and HPLC.Crystal data. C10H8N4O2S·2.875H2O, monoclinic, space group C2/c, a = 28.330(1), b = 12.851(1), c = 19.354(1) Å, β = 131.75(1)°, V = 5256.9(5) Å3, T = 130 K, Z = 16, λ = 0.71073 Å, R1 = 0.0647, wR2 = 0.1581 for 3237 independent reflections with I > 2σ(I). The water molecules are to a large extent disordered. Two water molecules, one in a general position and one on a 2-fold axis, have full occupancy. The remaining nine water molecules are disordered and have 0.5 (general position) or 0.25 (special position) occupancy, assigned based on consideration of a possible topology of the hydrogen-bond network.
Acknowledgements
This work was supported by Grant 3T09A 02615 from the State Committee for Scientific Research, the Republic of Poland.References
- A. Favre, in Bioorganic Photochemistry, ed. H. Morrison, Wiley, New York, 1990, vol. 1, p. 379 Search PubMed; A. Favre and Y. L. Dubreuil, New J. Chem., 1991, 15, 593 Search PubMed; A. Favre and J.-L. Fourrey, Acc. Chem. Res., 1995, 28, 375 Search PubMed.
- K. M. Meisenheimer and T. H. Koch, Crit. Rev. Biochem. Mol. Biol., 1997, 32, 101 Search PubMed.
- Z. Wang, I. Huq and T. M. Rana, Bioconjugate Chem., 1999, 10, 512 CrossRef CAS; A. Favre, C. Saintome, J.-L. Fourrey, P. Clivio and P. Laugaa, J. Photochem. Photobiol. B: Biology, 1998, 42, 109 Search PubMed.
- A. R. Lapucha, Synthesis, 1987, 256 CrossRef CAS.
- K. Kaneko, H. Katayama, T. Wakabayashi and T. Kumonaka, Synthesis, 1988, 152 CrossRef CAS.
- P. T. Jörgensen, E. B. Pedersen and C. Nielsen, Synthesis, 1992, 1299 CrossRef.
- J. M. Dzik, T. Kulikowski, Z. Zielinski, J. Ciesla, W. Rode and D. Shugar, Biochem. Biophys. Res. Commun., 1987, 149, 1200 CrossRef CAS.
- J. Fox, D. Van Praag, I. Wempen, I. L. Doerr, L. Cheong, J. E. Knoll, M. L. Eidinoff, A. Bendich and G. B. Brown, J. Am. Chem. Soc., 1959, 81, 178 CrossRef CAS.
- S. Y. Wang, in Photochemistry and Photobiology of Nucleic Acids, ed. S. Y. Wang, Academic Press, New York, 1976, vol. 1, pp. 326–351 Search PubMed.
- U. Niedballa and H. Vorbrüggen, J. Org. Chem., 1974, 39, 3660 CrossRef CAS.
- K. A. Watanabe, S. S. Saluja, B. A. Otter and J. J. Fox, Tetrahedron Lett., 1972, 3911 CrossRef CAS.
- A. Neville and D. Cook, Can. J. Chem., 1969, 47, 743.
- A. Albert and E. P. Serjant, The Determination of Ionisation Constants, Chapman and Hall, New York, 1984, p. 14 Search PubMed.
- R. S. Coleman and E. A. Kesicki, J. Am. Chem. Soc., 1994, 116, 11636 CrossRef CAS.
- D. E. Bergstrom and N. J. Leonard, Biochemistry, 1972, 11, 1 CrossRef CAS.
- J. Zemlicka, J. Smrt and F. Sorm, Collect. Czech. Chem. Commun., 1964, 29, 635 CAS.
- J. Asakura and M. J. Robins, J. Org. Chem., 1990, 55, 4928 CrossRef CAS.
- B. C. Pal, J. Am. Chem. Soc., 1978, 100, 5170 CrossRef CAS.
- S. L. Murrov, Handbook of Photochemistry, Marcel Dekker, New York, 1973, p. 162 Search PubMed.
Footnotes |
| † Crystal data have been deposited with the Cambridge Crystallographic Data Centre, CCDC reference number 162336. See http://www.rsc.org/suppdata/p1/b1/b108380e/ for crystallographic files in .cif or other electronic format. |
| ‡ Locants for the NMR data are those shown in the non-systematic numbering scheme shown in Scheme 1. |
| § DTE–DTT represents dithioerythritol–dithiothreitol. |
| This journal is © The Royal Society of Chemistry 2002 |