Cascade radical synthesis of heteroarenes via iminyl radicals†
W. Russell Bowman*, Colin F. Bridge, Philip Brookes, Martin O. Cloonan and David C. Leach
Department of Chemistry, Loughborough University, Loughborough, Leicestershire, UK LE11 3TU
First published on 5th December 2001
Abstract
A novel cascade cyclisation protocol has been developed which ‘zips up’ two rings to form new tetracycles. In the protocol, treatment of vinyl bromides and iodides with hexamethylditin (Me3Sn˙) yields intermediate vinyl radicals which undergo 5-exo cyclisation onto nitrile groups to yield intermediate iminyl radicals. The iminyl radicals can undergo 6-endo cyclisation (or 5-exo followed by neophyl rearrangement) to yield tetracyclic π-radicals which lose hydrogen (H˙) in an H-abstraction step. Methyl radicals, generated from the breakdown of trimethylstannyl radicals (Me3Sn˙), are proposed as a possible H-abstractor for this final oxidative step. The protocol has been used to synthesise the tetracyclic rings A–D (tetracycle indolizino[1,2-b]quinolin-9(11H)-one) of the anticancer alkaloids camptothecin, mappicine, nothapodytine B and nothapodytine A. The protocol has also been applied to the synthesis of analogues of camptothecin in which ring A has been replaced by thiophene (8,10-dihydrothieno[2′,3′:5,6]pyrido[2,3-a]indolizin-8-one) and ring D by pyrrole (10H-pyrrolizino[1,2-b]quinoline) and benzene (11H-indeno[1,2-b]quinolin-11-one).
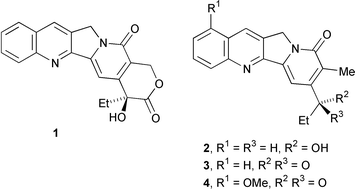
Camptothecin 1 and the related alkaloids, mappicine 2, nothapodytine B 3 and nothapodytine A 4, are important anticancer and antiviral alkaloids and several analogues are used as pharmaceuticals. The chemistry and pharmacology of camptothecins and analogues have comprehensively been reviewed.1 Camptothecin and mappicine have been popular targets of synthesis and a wide variety of protocols have been used.1 For instance, syntheses have included hetero Diels–Alder reactions,2 condensation to form the pyridone ring in the synthesis3 and even a biomimetic synthesis.4 Two radical protocols have been reported. Curran and co-workers have synthesised camptothecin and its analogues5–7 and mappicine8via bimolecular radical additions of radicals derived from 6-iodopyridin-2(1H)-ones onto areneisonitriles. A synthesis using radical cyclisation of quinolin-2-yl radicals onto pyridones has also been recently reported.9
We now report our studies aimed at the synthesis of camptothecin and mappicine. We have developed a new radical protocol for the synthesis of the tetracyclic heteroarenes, which includes rings A–D of camptothecin and mappicine, using [4 + 2] radical annulation via cyclisation of intermediate iminyl radicals, generated by vinyl radical cyclisation onto nitriles, onto arenes (see Scheme 1). The preliminary results of this research have been published.10
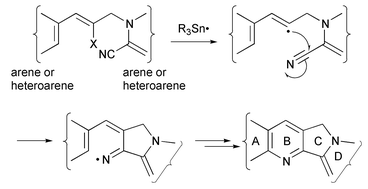 | ||
| Scheme 1 General synthetic protocol. | ||
Radical cyclisation is now commonly used for the synthesis of heterocycles and has become an important methodology.11 However, the synthesis of heteroarenes using radical cyclisation has been surprisingly little studied.11 A number of interesting examples of radical heteroarene synthesis have been published in recent years and include the synthesis of indoles,12 benzothiazoles,13 benzo[h]quinolines,14 benzothiophenes,15 pyridines,16,17 quinoxalines17 and camptothecin and analogues.5–8 Cascade radical cyclisations have also been applied to the synthesis of non-heteroarene polycyclic natural products.18 Radical cascade reactions facilitate ‘one-pot’ reactions which cut out purification at each step thereby facilitating higher overall yields and less waste. We were inspired to apply cascade radical reactions to the synthesis of heteroarenes by the examples of Curran and co-workers in their studies of radical synthesis of camptothecin which demonstrated the possibility for the synthesis of heteroarenes.5–8
Our radical studies have shown the utility of N-centred radicals in synthesis for the formation of C–N bonds. For instance, aminyl radicals, generated from sulfenamides19 or by cyclisation of C-centred radicals onto imines,20 have proved useful for the formation of C–N bonds and tandem cyclisations. The addition of C-centred radicals onto imines has been reviewed recently.21,22 The formation of C–N bonds using cyclisation of iminyl radicals has been successfully exploited by Zard.23 The rate of 5-exo cyclisation onto alkenes is one order of magnitude slower than for C-centred radicals.24 We have extended our studies to the cyclisation of C-centred radicals onto nitriles to generate intermediate iminyl radicals, which can undergo tandem cyclisations onto alkenes.25 However, the rate of cyclisation onto nitriles is slow and faster competing reactions need to be avoided in the design of syntheses. For example, the rate cyclisation of 4-cyanobutyl radicals [˙CH2(CH2)3CN] is slow (4 × 103 s−1 at 25 °C).26
In planning our synthetic protocol we were encouraged by reports in the literature which indicated that 6-endo cyclisation of iminyl radicals onto arenes (or 5-exo cyclisation followed by a neophyl rearrangement) was possible, thereby forming new pyridine rings by [4 + 2] radical annulation5–8,15,16 The proximity and orientation of the iminyl radicals to the arene rings and the driving force to aromatisation are important in facilitating these reactions. With these factors in mind we have developed a new protocol using vinyl radical cyclisation onto nitriles forming iminyl radicals which undergo cyclisation onto arenes. One example of this cascade sequence has previously been reported7 and several similar cyclisations onto nitriles and cyclisation of the resulting iminyl radicals have also been reported.5,16
With the synthesis of rings A–D of the alkaloids camptothecin 1, mappicine 2, nothapodytine B 3 (mappicine ketone) and nothapodytine A 4 as initial targets, we also wished to design a protocol that could be applied to related tetracycles with aromatic rings other than benzene and pyridone. The general synthetic protocol is shown in Scheme 1: generation of a reactive vinyl radical which undergoes 5-exo cyclisation onto the nitrile to yield an iminyl radical, which in turn undergoes 6-endo (or 5-exo followed by a neophyl rearrangement) and oxidation to yield the annulated pyridine ring, thereby ‘zipping up’ four rings in one pot.
Results and discussion
Synthesis of radical precursors for the synthesis of rings A–D of the alkaloids camptothecin 1, mappicine 2, nothapodytine B 3 and nothapodytine A 4
The aim was to plan a protocol that would allow facile synthesis of a range of precursors with different rings, i.e. by N-alkylation of the anion of a 2-cyano NH-heteroarene with a suitable vinylic bromide. In this procedure rings A and D could be varied. Initially we synthesised the precursors 13a–c required for the rings A–D of camptothecin and nothapodytine A 4 as shown in Schemes 2 and 3.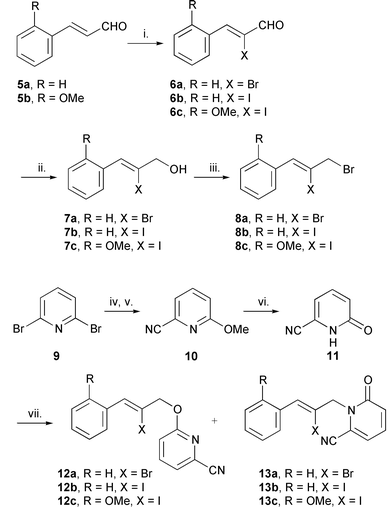 | ||
| Scheme 2 Reagents and conditions: i. Br2, Et3N, CH2Cl2 (6a, 90%); ICl, Et3N, CH2Cl2 (6b, 80%, 6c, 95%); ii. NaBH4, CeCl3, MeOH (7a, 100%), (7b, 100%), (7c, 85%); iii. CBr4, Ph3P, MeCN (8a, 100%, 8b, 100%, 8c, 80%); iv. NaOMe, MeOH, reflux; (73%); v. CuCN, DMF, reflux, 48 h (10, 62%); vi. TMSCl, NaI, MeCN (11, 82%); vii. 8a–c, NaH, LiCl, DME, DMF (13a, 71% from 8a, 13b, 63% from 8b, 13c, 45% from 8c). | ||
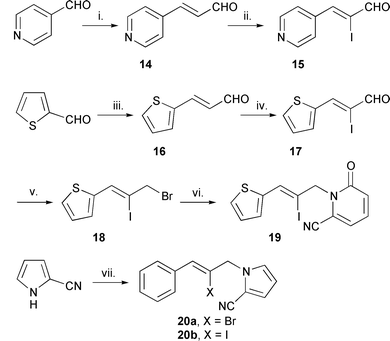 | ||
Scheme 3 Reagents and conditions: i. Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCHO (14, 83%); ii. ICl, Et3N, CH2Cl2 (15, <5%); iii. MeCHO, NaOH, EtOH, H2O (16, 49%); iv. ICl, Et3N, CH2Cl2 (17, 80%); v. NaBH4, CeCl3, MeOH; (93%); CBr4, resin-PPh3, MeCN; vi. 11, NaH, LiCl, DME, DMF (19, 21%, 18 not purified); vii. KOH, DMF, 8a and 8b (20a, 87%
from 8a), (20b, 96% from 8b). CHCHO (14, 83%); ii. ICl, Et3N, CH2Cl2 (15, <5%); iii. MeCHO, NaOH, EtOH, H2O (16, 49%); iv. ICl, Et3N, CH2Cl2 (17, 80%); v. NaBH4, CeCl3, MeOH; (93%); CBr4, resin-PPh3, MeCN; vi. 11, NaH, LiCl, DME, DMF (19, 21%, 18 not purified); vii. KOH, DMF, 8a and 8b (20a, 87%
from 8a), (20b, 96% from 8b). | ||
The cinnamaldehydes 5a and 5b were converted to the corresponding α-halogenocinnamyl bromides 8a–c in high yield by standard procedures (Scheme 2). The cinnamaldehydes 6a and 5b were commercially available. 2,6-Dibromopyridine 9 was converted to 6-oxo-1,6-dihydropyridine-2-carbonitrile 11 in overall good yields. The conversion of 2-cyano-6-methoxypyridine 10 to the cyanopyridone 11 was carried out using a literature procedure.5a We considered that the iodo analogue,5a 2-iodo-6-methoxypyridine, may give a higher yield but a similar yield of 6-methoxypyridine-2-carbonitrile 10 was obtained and hence did not provide a significant improvement. Direct conversion of 2-methoxypyridine to 2-cyano-6-methoxypyridine using butyllithium followed by toluene-p-sulfonyl cyanide27 had the advantage of one-step but gave a low yield (11%). Direct conversion of 2-bromo-6-oxo-1,6-dihydropyridine to 11 using the copper(I) cyanide method also failed.
The cyanopyridone 11 was alkylated with the α-halogenocinnamyl bromides 8a–c using the method of Curran and co-workers which favours N-regioselectivity.28 The radical precursors 13a and 13b were obtained in good yield [13a (71%), 13b (63%)]. The alkylation of 11 with 8c to yield 13c (45%) was not optimised. O-Alkylation was observed in each alkylation reaction; yields were 28 (12a), 25 (12b) and 26% (12c). Altering the reaction conditions did not improve the N/O regioselectivity, e.g. the use of greater equivalents of LiCl had little effect. The Z-stereochemistry of each radical precursor was determined by NOE difference NMR spectroscopy. The stereochemistry of the precursors is probably not important in the cyclisation steps because intermediate vinyl radicals rapidly interconvert between E- and Z-stereochemistry. However, there is no guidance in the literature to our knowledge on the effect of the E- and Z-stereochemistry on the initial halide abstraction step, and therefore, this effect of stereochemistry in these reactions is less clear. A possible alternative synthesis of the radical precursor 13a, involving the N-alkylation of 6-methoxypyridine-2-carbonitrile 10 with activated halides,29 is not viable as the pyridine ring failed to alkylate with benzyl bromide. This result can be rationalised in terms of the reduced nucleophilicity of the sp2 hybridised nitrogen in the pyridine ring due to the presence of the electron withdrawing nitrile functional group.
3-(Pyridin-4-yl)prop-2-enal 14 (E ∶ Z = 19.5 ∶ 1) was synthesised using a Wittig reaction involving pyridine-4-carbaldehyde and (formylmethylene)triphenylphosphorane30 in an 83% yield (Scheme 3). However, the iodination of 14 with iodine monochloride (ICl) resulted in decomposition with only traces of the required vinyl iodide 15. The decomposition can be expounded by the destabilisation of the iodo-bridged carbocation intermediate, relative to the phenyl analogue, by the electron withdrawing pyridine ring. Thus, this experiment points to a limitation of the protocol. (E/Z)-3-(2-Thienyl)prop-2-enal (β-2-thienylacrolein) 16 was synthesised using an aldol reaction involving thiophene-2-carbaldehyde and acetaldehyde in a 49% yield (E ∶ Z, 8.7 ∶ 1) and successfully iodinated (Scheme 3).31 The thiophene ring proved compatible with the chosen synthetic protocol, however, purification of 2-[(Z)-3-bromo-2-iodoprop-1-enyl]thiophene 18 by chromatography was not possible due to rapid hydrolysis to the corresponding alcohol. The chloride derivative also proved to be labile and the acetate which was stable could not be alkylated. Triphenylphosphine bound to a resin provided the bromide in reasonable purity and the alkylation step was carried out immediately due to the instability of the allyl bromide 18. The precursor 19 was obtained in a 21% yield and not optimised. The Z-configuration was confirmed from NOE difference 1H NMR spectroscopy. Only the N-alkylated product 19 proved stable and the O-alkylated isomer decomposed to a black polymer as observed in the decomposition of 18. This behaviour, as in the lability of the allyl bromide 18, can be related to the greater electron density of the thiophene ring relative to the phenyl ring. The lone pair of the sulfur atom aids the heterolytic fission of the carbon–bromide bond. Thus, the more electron rich 5-membered rings such as pyrroles and furans are predicted to be more problematic in this synthetic protocol.
Radical precursors with a five-membered heteroarene for ring D, 1-(2-halo-3-phenylprop-2-enyl)-1H-pyrrol-2-yl cyanides 20a and 20b, were also synthesised in high yields [20a (87%), 20b (96%)] by alkylation of 2-cyanopyrrole with the cinnamyl bromides 8a and 8b using potassium hydroxide in DMF (Scheme 3).
Radical cyclisation reactions: synthesis of rings A–D of the alkaloids camptothecin 1, mappicine 2, nothapodytine B 3 and nothapodytine A 4
The cascade radical cyclisations using the radical precursors 13a–c were carried out under a range of conditions using hexamethylditin to generate the intermediate trimethyltin radicals (Me3Sn˙) (Scheme 4). The purpose of using hexamethylditin as opposed to tributyltin hydride is to avoid the formation of reduction products. The reactions were heated for 48 h at 150 °C and irradiated with sunlamps (2 × 150 W) to facilitate cleavage of the Sn–Sn bond in hexamethylditin. The solutions required rigorous removal of oxygen by use of the freeze–thaw technique with nitrogen gas. The bromo precursor 13a was examined initially which gave a 20% yield of the tetracycle indolizino[1,2-b]quinolin-9(11H)-one 29a. The starting material was also recovered in an 80% yield indicating clean conversion of the precursor to product. Insoluble polymeric organotin material deposited in the flask in all reactions but did not affect work-up or purity of products. We studied the use of alternative solvents because of the known ability of reactive radicals to add to arenes. A change to aliphatic solvents had a minimal effect on the product yield. Acetonitrile has proved useful in radical radicals, therefore, reactions in butyronitrile and valeronitrile were studied but resulted in only 10 and 31% yield of 29a respectively with a large degree of substrate decomposition in the latter solvent. Similarly, tert-butyl alcohol has proved useful (no α-hydrogens which can easily be abstracted). However, high boiling point tertiary alcohols such as 2-methylhexan-2-ol also exhibited a high degree of substrate decomposition. These results exclude a solvent rationale for the poor conversions observed.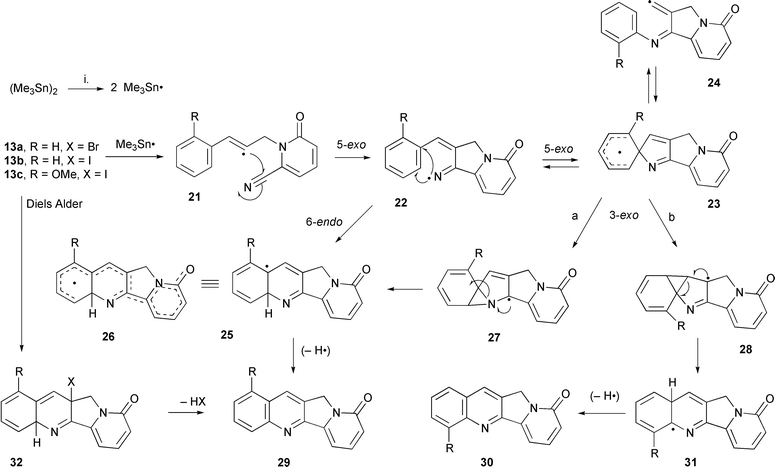 | ||
| Scheme 4 Reagents and conditions: i. (Me3Sn)2, t-BuPh, 150 °C (29a, 73%, 29c 45%; 21–31, a, R = H; c, R = OMe). | ||
Thus we examined the iodo derivative 13b as a synthetic equivalent based on the fact that the vinyl–iodo bond is much weaker than the vinyl–bromo bond. The radical reaction using the iodo radical precursor 13b under the same reactions conditions gave the tetracycle indolizino[1,2-b]quinolin-9(11H)-one 29a in higher yields. Optimised conditions [hexamethylditin (7.9 equiv.), tert-butylbenzene, 150 °C, 48 h, sunlamp irradiation, under nitrogen] gave 73% of 29a with some unaltered starting material 13b (15%). Large amounts of organotin polymers were also observed, especially if the reaction was carried out at temperatures higher than 150 °C. Longer reaction times used up the starting material but did not increase the yield of 29a. Thus the difference in yield between the bromo and iodo precursors can be related to the difference in bond strengths, although the exact role of steric and conformational effects, if any, are unclear. The cascade radical cyclisation using the methoxy precursor 13c was also carried out to synthesise the A–D ring skeleton [1-methoxyindolizino[1,2-b]quinolin-9(11H)-one 29c] of the anticancer alkaloid nothapodytine A 4. The same reaction conditions were used and only one isomer 29c was obtained in 45% yield (unoptimised) (Scheme 4). Interestingly, none of the possible isomer 30c was detected which suggested that the cyclisation was proceeding via 6-endo cyclisation and not via a spiro intermediate 23c. Conclusive evidence for the formation of the 1-methoxy isomer was obtained from NOE difference 1H NMR spectra and from the 13C NMR spectrum.
The full mechanism of the radical cascade reaction is not clear. Hexamethylditin cleaves under the influence of heat and/or light to yield trimethyltin radicals which abstract the iodine/bromine atom from the vinyl precursors 13a–c to yield the intermediate vinyl radical 21. The reactive vinyl radical undergoes 5-exo cyclisation onto the nitrile generating the iminyl radical 22. In the absence of faster competing reactions the attack on the nitrile is favoured. 5-exo Cyclisation onto nitriles by reactive aryl or vinyl radicals to form iminyl radical intermediates is well documented.22,23,25 In the presence of a hydrogen source (i.e. Bu3SnH) the iminyl intermediates can be trapped by reduction, ruling out a one-step concerted reaction32 (see Scheme 7). The resulting iminyl radical intermediate 22 undergoes cyclisation onto the aryl ring by either a 6-endo or a 5-exo cyclisation (Scheme 4). The 6-endo cyclisation generates the π-radical 25 which is oxidised to the tetracycle 29. The π-radical 25 is stabilised by delocalisation of the unpaired electron over almost all of the structure as represented in 26. The stability of this radical may present a driving force for 6-endo cyclisation or formation in a neophyl rearrangement from the 5-exo cyclised intermediate 23.
The observation of rearranged products reported in the literature,5–8 as well as stereoelectronic arguments, in cyclisation reactions of vinyl and iminyl radicals onto arenes (i.e. 5-exo cyclisation preferred to 6-endo cyclisation) necessitates the consideration of a spirodiene intermediates such as 23. Direct evidence for the involvement of the cyclohexadienyl radicals in cyclisations onto arenes has arisen from experiments by Hey33 and Crich34 using radical traps. A neophyl rearrangement generates the stable aromatic π-radicals 25 and/or 31. This 1,2-shift occurs by an initial 3-exo-trig process as proposed by Curran on similar systems.5–8 Cyclisation by pathway a (Scheme 4) onto the iminyl double bond generates the unrearranged product 29 whereas cyclisation onto the vinyl bond by pathway b gives 30. For the reactions of precursors 13a and 13b the product will be the same. However, for the methoxy precursor 13c both products (29c and 30c) could be predicted but only the 1-methoxyindolizino[1,2-b]quinolin-9(11H)-one 29c was formed. Radical 27 would be predicted to be more stable than 28 due to delocalisation across the pyridone and the allyl bond as well as its captodative structure. Radical 28 has only stabilisation by the iminyl group. The lack of a rearranged tetracycle for the methoxy system 13c is consistent with the relative stabilities but provides no conclusive evidence for the existence of the spirodiene 23. Semi-empirical AM1 calculations by Nanni et al.17 predict ring opening of spiro intermediates. Thus a rearranged product could arise from opening of the spirodiene to a vinyl radical 24. However, in this case it would seem less thermodynamically favoured than in the iminyl system studied by Nanni.17 No evidence was obtained for a 6-endo-ipso cyclisation and subsequent loss of an OMe radical.
An iodine atom transfer mechanism in which hexamethylditin is only required in sub-equivalent amounts as an initiator is unlikely because abstraction of iodine from the starting vinyl iodide by the stable π-radical 25 is extremely energetically unfavourable and hence can be ruled out. Homolytic cleavage of the vinyl–iodine bond is unlikely to initiate the radical reaction. A blank reaction with 13b without the addition of hexamethylditin gave unaltered starting material (99%) with only traces of the tetracycle 29a. This may reflect a more diverse system involving a Diels–Alder cycloaddition as a minor pathway to the tetracycle. Cycloadduct 32 can in theory lose HX via a 1,4-conjugate elimination or a radical mechanism to generate 29a (Scheme 4).
The mechanism of the oxidative step, 25 to 29, is unclear. The most likely route is abstraction of the 4a-hydrogen but the identity of the H-abstractor is unknown. There is also a strong driving force towards aromatisation of 25 to yield the product 29 which makes H-abstraction favourable. Disproportionation as a major route can be ruled out because no dihydro products were obtained and the yields for 29a were higher than 50%. The mechanism of oxidation is discussed by Curran and co-workers but with no conclusion.5–8 The mechanism of oxidative cyclisations with Bu3SnH has been an area of debate but the prevailing view at present is that the azo initiators such as AIBN play a major role35 and not a radical anion SRN1 type mechanism as we initially postulated.36,37 These considerations are not relevant here because of the absence of such initiators. If H-abstraction is the means of ‘oxidation’ what is the H-abstractor? Traces of oxygen will rapidly react with trimethyltin radicals to yield peroxy radicals (Me3SnOO˙)38 which are predicted to be good H-abstractors. We believe that this possibility can be largely ruled out because the reactions were very carefully deoxygenated using freeze–thaw techniques. Large amounts of organotin polymer were observed in all the reactions which originates from the breakdown of Me3Sn˙ radicals (Scheme 5).39 The identity of the organotin polymer is unknown and it was not characterised but we assume that it is likely to be 34, which is known to result from the breakdown of hexamethylditin via Me3Sn˙ radicals and insertion of dimethylstannylene 33 into the Sn–Sn bond.39 The methyl radical, generated by breakdown of the Me3Sn˙ radical to methyl radical and dimethylstanylene 33, is an obvious H-abstractor (Scheme 5).
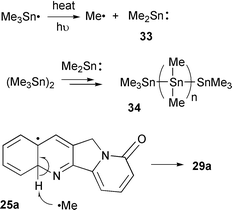 | ||
| Scheme 5 Putative mechanism of hydrogen abstraction. | ||
We investigated the use of di-tert-butyl peroxide to generate Me3Sn˙ radicals more efficiently and also at a lower temperature and time with the aim of largely preventing the problem of the organotin polymer formation (Scheme 6). tert-Butoxyl radicals rapidly cleave the Sn–Sn bond providing Me3Sn˙ radicals at a lower temperature and also efficiently abstract the 4a-hydrogen from the π-radical intermediate 25a (Scheme 6). Various equivalents of di-tert-butyl peroxide and hexamethylditin and reaction conditions were studied using a sealed Schlenk tube for the reaction to prevent loss of the volatile peroxide (bp 109 °C). The reaction mixtures were carefully deoxygenated with nitrogen using the freeze–thaw technique. The optimum conditions for the conversion of the iodo precursor 13b to the tetracycle 29a were (t-BuO)2 (1.7 equiv.), (Me3Sn)2 (2.2 equiv.) and 120 °C for 24 h under nitrogen in a sealed Schlenk tube. These conditions gave 29a in 60% yield and unaltered starting material 13b (10%). The yield of the tetracycle from the bromo precursor 13a [29a (18%) and unaltered 13a (81%)] was similar to the reaction with hexamethylditin alone. However, when 13a was reacted with more of both reagents and for a longer reaction time, the yield of 29a increased to 50% and no starting material 13a was detected [(t-BuO)2 (8.6 equiv.), (Me3Sn)2 (7.9 equiv.), 120 °C, 48 h]. When the reactions were attempted using di-tert-butyl peroxide without hexamethylditin, extensive decomposition took place. This procedure has good potential for cleaner and faster reactions and is being further investigated. However, this protocol did not work in the presence of methoxy substituents. Precursor 13c gave only extensive decomposition and no tetracycle 29c was detected. This result can be viewed in terms of hydrogen abstraction from the OMe group by the highly electrophilic tert-butyl peroxide radical.
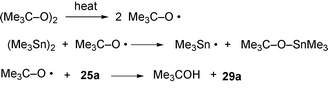 | ||
| Scheme 6 Putative mechanism of hydrogen abstraction by tert-butoxyl radicals. | ||
Reports in the literature indicate that the use of acetone as a triplet sensitiser facilitates cleavage of the Sn–Sn bond in triplet hexamethylditin at a much lower temperature.40 We investigated this protocol with the aim of improving yields and cutting down on the formation of organotin polymers. This protocol gave 29a in 19% yield with unaltered 13a (40%) [acetone, (Me3Sn)2 (10 equiv.), 350 nm UV irradiation, 4 days]. Yields could not be improved either by using higher equivalents of acetone or hexamethylditin and/or longer reaction times. However, it is interesting to note that the tandem cyclisation takes place at room temperature.
Attempted tandem radical cyclisation reaction using an aliphatic nitrile 35a
We were interested to determine the scope of the cyclisation protocol and attempted the tandem cyclisation on an aliphatic nitrile. Just one example of a similar reaction has been reported.27 The aliphatic nitrile precursor 35a was synthesised by alkylation of the anion of diethyl 2-(cyanomethyl)propanedioate with 8a. The precursor 35b was synthesised using a Mitsunobu reaction between 7b and diethyl 2-(cyanomethyl)propanedioate in high yield. Treatment of 35a with Bu3SnH (syringe pump addition) and AMBN [azobismethylisobutyronitrile, or by IUPAC nomenclature, 2-(1-cyano-1-methylpropylazo)-2-methylbutyronitrile] in refluxing cyclohexane gave the ketone 39 in a 32% yield (Scheme 7). AMBN was used because it is partly soluble in refluxing cyclohexane whereas AIBN is not. Reduction of the iminyl radical 37 by Bu3SnH results in the formation of imine 38 which hydrolyses on work up to the ketone 39. The intermediate iminyl radical reacts faster with the Bu3SnH than cyclisation onto the phenyl ring. Similar behaviour is predicted for the other radical precursors, e.g.13a–c, but has not yet been tested. Further reactions using hexamethylditin under similar conditions which had proved successful for precursors 13a–c, but without the large excess of hexamethylditin, were unsuccessful and gave only starting material or decomposed materials. Interestingly, only recovered starting material and/or decomposition was observed for the bromo derivative 35b. The reasons for the failure of the tandem reaction are not clear.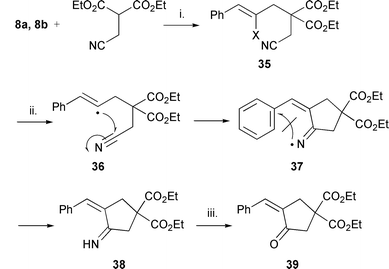 | ||
| Scheme 7 Reagents and conditions: i. diisopropyl azodicarboxylate, Ph3P, dioxane, rt, 70 h; 31 (97%); ii. Bu3SnH, AMBN, cyclohexane, 4 h, reflux; iii. water in the work-up (39, 32%) (a, X = Br; b, X = I). | ||
Synthesis of the thieno[3,2-b]pyridine ring system 44 using radical cyclisation
The thiophene analogue 19 showed similar cyclisation behaviour to the phenyl precursors 13 (Scheme 8). Using the sunlamp and hexamethylditin conditions at 150 °C, 8,10-dihydrothieno[2′,3′:5,6]pyrido[2,3-a]indolizin-8-one 44 was isolated in a 60% yield with 8% of the starting material recovered. The presence of the thieno[3,2-b]pyridine ring system (in 44) and not the thieno[2,3-b]pyridine system (in 46) was confirmed by NOE difference 1H NMR spectroscopy. An enhancement was observed between 2-H and the 3-H with no enhancement between 11-H and 2-H or 3-H of the thiophene ring. The 11-H also showed an enhancement only with 10-H. Further evidence of the structure came from 1H and 13C NMR spectroscopic comparison with the literature data of the thiophene analogue of camptothecin, A-nor-9-thia-20(RS)-camptothecin, which has the same tetracyclic skeleton as the product 44. This thiophene analogue of camptothecin has been found to be less active against cancer cells.41 The formation of the thieno[3,2-b]pyridine ring system and not the rearranged product, thieno[2,3-b]pyridine system in 46, can be rationalised by the same arguments put forward for the methoxy precursor 13c, i.e. cyclisation of 41 onto the imine to yield the more stable radical intermediate 43 is more favoured than cyclisation onto the alkene to yield 45. Interestingly, no products were observed which were consistent with ring opening of the spirodiene 41 to yield a ring opened sulfanyl radical. A related pathway has been postulated to occur in dihydrofuran spirocycles.42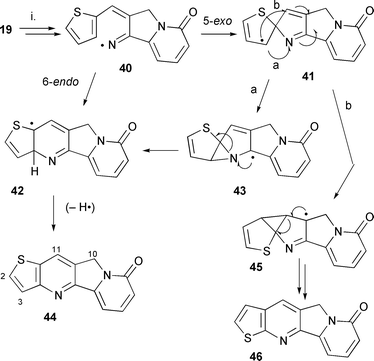 | ||
| Scheme 8 Reagents and conditions: i. (Me3Sn)2, t-BuPh, 150 °C, 48 h (44, 60%). | ||
Synthesis of 10H-pyrrolizino[1,2-b]quinoline 49 using radical cyclisation
The pyrrole system 20 was investigated to determine the suitability of azole rings for ring D in the cascade reaction. The cyclisation also proved an instructive probe of the transition state of this tandem cyclisation (Scheme 9). The reaction yielded the desired product, 10H-pyrrolizino[1,2-b]quinoline4349, in a 3% yield from the iodo substrate 20b using the hexamethylditin and di-tert-butyl peroxide conditions. The radical precursor 20b was recovered in a 28% yield and decomposition was the dominant event in the reaction. Application of the sunlamp and hexamethylditin conditions at 150 °C also gave predominately decomposition with 19% starting material recovered with none of the tandem cyclised product observed. The bromo substrate 20a was less reactive as expected resulting only in decomposition but with a greater recovery of 20a. In the initial 5-exo cyclisation of the vinyl radical intermediate 47 to the iminyl radical 48, strain in the cyclisation onto the nitrile due to the formation of a 5,5-membered bicycle is consistent with a less efficient cyclisation. The involvement of an aromatic system in this bicyclic generating step would be expected to augment this strain as opposed to a saturated 5-membered ring. The difficulty in generating bicyclic 5-membered rings is well documented in radical cyclisations onto imidazoles, pyrroles and benzoimidazoles.36,44 Zanardi et al.45 have also reported similar reactivity in a cascade sequence of reactions which include a 1,5-cyclisation onto a nitrile. Thus the synthetic usefulness of our protocol is restricted to the use of 5-membered rings for ring D of the tetracycle by strain in the initial 5-exo cyclisation step.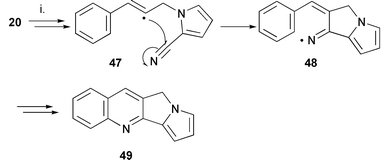 | ||
| Scheme 9 Reagents and conditions: i. (Me3Sn)2, t-BuPh, 150 °C, 48 h (49, 3%). | ||
Synthesis of the 11H-indeno[1,2-b]quinolin-11-one systems 52 and 53 and studies of the intermediacy of spirodienyl radicals
The intermediacy of the spirodienyl radicals (e.g.23, 41) is likely but not proven and is important if substituted arenes are to be used in the protocol. The synthesis of the tetracycle 29c, rings A–D of nothapodytine A, gave only one isomer suggesting 6-endo cyclisation rather than 5-exo cyclisation, followed by a neophyl rearrangement. However, the selectivity towards isomer 29c as opposed to 30c could be explained as previously mentioned by intermediate radical stability of 27c, formed in the 3-exo cyclisation of the π-radical intermediate 23c to 27c. Reversibility between the spirodiene 23c and the iminyl radical intermediate 22c is possible but ring opening of 23c to an alternative vinyl radical 24 is energetically unlikely. There is evidence in the literature for both 6-endo cyclisation17 and for 5-exo cyclisation followed by a neophyl rearrangement.6–8 In related cyclisations onto arenes by vinyl radicals, arenes with o-fluorine substituents5c and a range of m- and p-substituents give both regioisomers5c,6,8 indicating 5-exo cyclisations, followed by neophyl rearrangements to both isomers. Based on the argument for selective formation of 27 from the spirodiene the logical deduction would be that stabilisation of the 3-exo pathway onto the olefin should increase the probability of formation of the ‘rearranged’ product(s). This argument can be explored by replacing the methylene group with the more efficient radical stabilising carbonyl group, assuming its position would have greater effect on the 3-exo cyclisation onto the olefin than the imine. The precursor chosen to explore this deduction, to prove the existence of the spirodiene in these systems and to explore the factors that govern its behaviour was 2-[(Z)-2-iodo-3-(3-methoxyphenyl)prop-2-enoyl]benzonitrile 51. Furthermore, this experiment also explored the synthetic versatility of our radical protocol to using ring D arenes. The substrate was synthesised in three high yielding steps from the commercially available 2-bromoacetophenone and 3-methoxybenzaldehyde (Scheme 10). The radical precursor 51 gave off some iodine on silica gel chromatography and therefore fast flash chromatography was used in the purification step.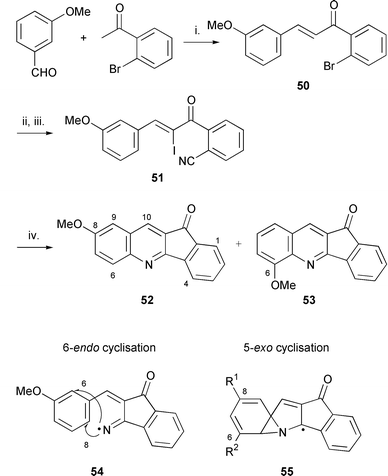 | ||
| Scheme 10 Reagents and conditions: i. NaOH, EtOH, rt, 24 h (83%); ii. CuCN, DMF, 150 °C, 4 h (63%); ICl, Et3N, CH2Cl2 (100%); iii. (Me3Sn)2, toluene, sunlamp irradiation (2 × 150 W), 80 °C, 24 h (52 ∶ 53 = 1 ∶ 3.2, 25%). | ||
Interestingly, the cascade reaction of 51 can be carried out under much milder conditions than the previous systems. This is consistent with a weaker vinyl–iodine bond due to the presence of the carbonyl group. The 11H-indeno[1,2-b]quinolin-11-one systems 52 and 53 were synthesised in a combined yield of 25% using the sunlamp and hexamethylditin protocol at 80 °C in toluene (Scheme 10). The yield was not optimised but no other detectable products were formed. The reaction was achieved using one equivalent of hexamethylditin. Two isomers were formed in a ratio of 6-methoxy 53 ∶ 8-methoxy 52 = 3.2 ∶ 1. The major isomer was shown to be the 6-methoxy isomer 53 by NOE difference spectra. The structure of the 8-methoxy minor isomer 52 was determined by NOE studies and by agreement of the spectroscopic data with the literature data.46 Both compounds can be rationalised without invoking a spirodiene intermediate from a 6-endo pathway (see radical intermediate 54 in Scheme 10). There is no obvious reason for the preference for cyclisation of the iminyl radical 54ortho to the methoxy group which generates the major isomer 53 over cyclisation para to the methoxy group which gives the minor 8-methoxy isomer 52. The 1H NMR spectrum and GCMS of the purified mixture of isomers also indicated traces of two other isomers. This can only be explained by the involvement of a spiro radical intermediate. However, if a spiro intermediate is involved in the major pathway, the preferred 3-exo cyclisation in the neophyl rearrangement is still onto the imine group rather than onto the vinyl group (β-position of the α,β-unsaturated ketone) to give a stabilised intermediate 55 as shown in Scheme 10 (55, R1 = OMe, R2 = H for 6-methoxy product 53 and 55, R1 = H, R2 = OMe for 8-methoxy product 52). Cyclisation onto the vinyl group would yield the 7- and 9-methoxy isomers. In summary, we have no conclusive evidence to prove that all products arise solely from a spiro intermediate.
At higher temperatures (150 °C) and equivalents of hexamethylditin, the secondary alcohol derivatives of 52 and 53 were also isolated. This is best explained by initial formation of the indeno[1,2-b]quinolin-11-one heterocycles 52 and 53 followed by reduction of the carbonyl group under the radical conditions. The similarity of the yields and isomeric ratio with the above 11H-indeno[1,2-b]quinolin-11-one systems 52 and 53 provides evidence for this mechanism. The yield of the latter reaction was not optimised but does show that a benzonitrile moiety can be successfully introduced into the structural framework of the general protocol outlined in Scheme 1.
In conclusion, our protocol has given good yields of the rings A–D of the skeleton of camptothecin and nothapodytine A and provides a facile synthetic route to a range of other tetracyclic analogues as well as elucidating some of the main features of the reaction mechanism that will guide future rational design.
Experimental
Commercial dry solvents were used in all reactions except for light petroleum and ethyl acetate which were distilled from CaCl2 and dichloromethane (CH2Cl2) was distilled over phosphorus pentaoxide. Light petroleum refers to the bp 40–60 °C fraction. Sodium hydride was obtained as 60% dispersion in oil and was washed with light petroleum. Mps were determined on an Electrothermal 9100 melting point apparatus and are uncorrected. Elemental analyses were determined on a Perkin Elmer 2400 CHN Elemental Analyser in conjunction with a Perkin Elmer AD-4 Autobalance. IR spectra were recorded on a Perkin-Elmer Paragon 1000 FT-IR spectrophotometer on NaCl plates. 1H (250 MHz) and 13C (62.5 MHz) NMR spectra were recorded on a Bruker AC-250 spectrometer as solutions of CDCl3 with tetramethylsilane (TMS) as the internal standard for 1H NMR spectra and deuteriochloroform the standard for 13C NMR spectra unless otherwise specified. Chemical shifts are given in parts per million (ppm) and J values in hertz (Hz). Mass spectra were recorded on a JEOL SX102 mass spectrometer or were obtained by the EPSRC Mass Spectrometry Service at University of Wales, Swansea. GC-MS was carried out on Fisons 8000 series GC-MS using a 15 m × 0.25 mm DB-5 column and an electron impact low resolution mass spectrometer. TLC using silica gel as absorbent was carried out with aluminium backed plates coated with silica gel (Merck Kieselgel 60 F254). Silica gel (Merck Kieselgel 60 H silica) was used for column chromatography unless otherwise specified.General procedure for halogenations of α,β-unsaturated aldehydes and ketones
O), 134.5 (4′-C), 129.0, 130.8, 132.9, 156.2 (Ph ∶ 2-, 3- and 4-C, 3-C); m/z 258 (M+, 39%), 131 (26), 130 (30), 127 (12), 103 (92), 91 (45), 77 (100) and 51 (42). The spectral data were in agreement with those reported in the literature.48General procedure for the reduction α,β-unsaturated aldehydes
General procedure for bromination of alcohols
) and 131.85 (5-C); m/z (EI) 328 (M+, 35%), 248 (100) and 121 (100).General procedure for cyanations
General procedure for the alkylation of 6-oxo-1,6-dihydropyridine-2-carbonitrile 11
Elution using a gradient mixture of dichloromethane–ethyl acetate (10 ∶ 1–5 ∶ 1 v/v) gave (Z)-1-(2-bromo-3-phenylprop-2-enyl)-6-oxo-1,6-dihydropyridine-2-carbonitrile 13a (1.11 g, 71%), mp 100–101 °C (from dichloromethane–light petroleum) [Found (M + H)+, 315.0128. (C15H11BrN2O + H) requires 315.0133] (Found: C, 57.15; H, 3.4; N, 8.9. C13H11BrN2O requires C, 57.2; H, 3.5; N, 8.8%); νmax(CH2Cl2)/cm−1 3054, 2233, 1671, 1595, 1536, 1431, 1265, 1145, 807, 737 and 697; δH 5.10 (2 H, s, CH2N, NOE ds, enhancement from vinylic-H, 6.5%), 6.66–6.68 (1 H, m, 5-H or 3-H, pyridone), 6.76–6.78 (1 H, m, 5-H or 3-H, pyridone), 7.05 (1 H, s, vinylic-H, NOE ds, enhancement from CH2N, 7.9%), 7.19–7.30 (4 H, m, overlap of pyridone 4-H and Ph-H) and 7.56–7.58 (2 H, m, Ph-H; NOE ds, enhancement from vinylic H, 7.7%); δC 55.60 (CH2N), 113.21 (vinyl α-C), 118.06 (CN), 121.52 (2-C), 116.78, 127.79, 128.69, 129.07, 129.49, 132.81 and 138.49 (Ph: 2/6-, 3/5- and 4-C, vinyl β-C, 3-C, 4-C and 5-C), 134.82 (Ph: 1-C) and 160.92 (6-C); m/z (EI) 235 (20%), 116 (15), 115 (100), 103 (8), 89 (10), 77 (8), 63 (8), 51 (10) and 39 (20).
General procedure for the alkylation of 2-cyanopyrrole
Radical cyclisations
9,11-Dihydroindolizino[1,2-b]quinolin-9-one 29a48. A solution of (Z)-1-(2-iodo-3-phenylprop-2-enyl)-6-oxo-1,6-dihydropyridine-2-carbonitrile 13b (170 mg, 0.476 mmol) in tert-butylbenzene (6.5 cm3) in a flat two-necked flask (pyrex, wall thickness 1 mm) was purged with nitrogen for 20 min under a reflux condenser connected to a nitrogen bubbler. Hexamethylditin (1.2 g, 3.66 mmol) was added by syringe and the resultant solution purged with nitrogen for a further 30 min. The mixture was irradiated with sunlamps (2 × 150 W) at 150 °C for 48 h. The reaction was cooled to ambient temperature, diluted with methanol (50 cm3) and evaporated under reduced pressure. The residue was dissolved in methanol (2 cm3) was placed on a silica gel column. Elution with dichloromethane removed the solvent tert-butylbenzene. A mixture of dichloromethane–ethyl acetate (5 ∶ 1 v/v) eluted 13b (25 mg, 15%). Elution using a mixture of ethyl acetate–methanol (6 ∶ 1 v/v) gave 29a (80 mg, 73%), mp 253–254 °C (from ethanol) (lit.,48 265 °C) [Found (M + H)+, 235.0870. C15H10N2O + H) requires 235.0871] (Found: C, 76.5; H, 4.1; N, 11.9. C15H10N2O requires C, 76.9; H, 4.3; N, 11.9%); νmax(Nujol)/cm−1 1659 (CO, pyridone), 1621, 1614, 1592, 1566, 1554, 1532, 1153, 1073, 967 and 720; δH 5.23 (2 H, s, 11-H), 6.72 (1 H, d, J 9.2, 8-H), 7.27 (1 H, d, J 6.8, 6-H), 7.59–7.68 (2 H, m, 2-H and 7-H), 7.77–7.81 (1 H, m, 3-H), 7.88 (1 H, d, J 8.0, 1-H), 8.18 (1 H, d, J 8.8, 4-H), 8.31 (1 H, s, 12-H); δC 49.91 (11-C), 100.88 (6-C), 120.53 (8-C), 127.66 (2-C), 128.07 (1-C), 128.02 and 128.57 (11a-C and 12a-C), 129.65, 130.33 and 130.84, (3-C, 4-C and 12-C), 140.36 (7-C), 146.13 and 148.83 (4a-C and 5b-C), 152.97 (5a-C) and 161.55 (9-C); m/z (EI) 234, 205, 178, 151, 140, 128, 115, 105, 91, 75, 63, 55, 49 and 43.
1-Methoxy-9,11-dihydroindolizino[1,2-b]quinolin-9-one 29c. The general procedure using 1-[2-iodo-3-(2-methoxyphenyl)prop-2-enyl]-6-oxo-1,6-dihydropyridine-2-carbonitrile 13c gave unaltered 13c (1.4%) and 1-methoxy-9,11-dihydroindolizino[1,2-b]quinolin-9-one 29c (45%), mp 252–253 °C (from dichloromethane–light petroleum) [Found (M + H)+, 265.0973. (C16H12N2O2 + H) requires 265.0976]; νmax(Nujol)/cm−1 1659, 1618, 1599, 1572, 1532, 1253, 1233, 1187, 1166, 1114, 1059, 1033, 987, 896, 850, 795 and 723; δH 4.02 (3 H, s, OMe), 5.14 (2 H, s, 11-H), 6.70 (1 H, d, J 9.0, 8-H), 6.88 (1 H, d, J 7.6, 2-H, NOE ds, enhancement from OMe, 3.8%), 7.21 (1 H, d, J 6.8, 6-H), 7.66–7.62 (2 H, m, 3-H and 7-H overlap), 7.71 (1 H, d, J 8.5, 4-H) and 8.66 (1 H, s, 12-H, NOE ds, enhancement from OMe, 0.4%); δC 50.08 (11-C), 55.88 (OMe), 100.84 and 105.14 (2-C, 6-C), 120.36 and 121.56 (4-C, 8-C), 120.49 (12a-C), 125.59 (3-C), 127.60 11a-C), 130.27 (12-C), 140.29 (7-C), 146.18 and 149.48 (4a-C, 5b-C), 152.92 (5a-C), 155.11 (1-C) and 161.52 (9-C); m/z (EI) 264 (M+, 100%), 249 (90), 235 (5), 221 (30), 205 (5), 192 (30), 166 (8), 140 (8), 110 (8), 96 (8), 82 (10), 75 (10), 63 (8) and 51 (8).
8,10-Dihydrothieno[2′,3′:5,6]pyrido[2,3-a]indolizin-8-one 44. (Z)-1-[2-Iodo-3-(2-thienyl)prop-2-enyl]-6-oxo-1,6-dihydropyridine-2-carbonitrile 19 (8%) and 8,10-dihydrothieno[2′,3′:5,6]pyrido[2,3-a]indolizin-8-one 44 (60%) (Found M+ 240.0357. C13H8N2OS requires 240.0357); νmax(CH2Cl2)/cm−1 1667, 1611, 1578, 1570, 1543, 1378 and 1105; δH 5.23 (2 H, s, 10-H, NOE ds, enhancement from 11-H), 6.69 (1 H, dd, J 9.0, 1.0, 5-H), 7.20 (1 H dd, J 7.0, 1.0, 7-H), 7.64–7.68 (2 H, m, 3-H, 6-H, overlap), 7.90 (1 H, d, J 5.6, 2-H), 8.42 (1 H, s, 11-H, NOE ds, enhancement from 10-H); δC 50.00 (10-C), 99.98 (5-C), 119.58 (7-C), 124.83 and 125.72 (2-C, 3-C), 127.38 and 134.26 (10a-C, 11a-C), 132.53 (11-C), 140.51 (6-C), 146.65 and 150.70 (3a-C, 4b-C), 157.02 (4a-C) and 161.60 (8-C); m/z (EI) 240 (M+, 65%), 226 (25), 211 (25), 138 (100), 95 (80), 83 (20), 67 (30), 57 (30) and 39 (27). The NOE difference spectra confirmed the thieno[3,2-b]pyridine ring system (i.e. the nitrogen and sulfur are trans to each other) and not the thieno[2,3-b]pyridine system (the nitrogen and sulfur are cis). An enhancement was observed between 2-H and 3-H with no enhancement between 11-H and 3-H or 2-H. 11-H showed an enhancement only with 10-H. 3-H showed an enhancement only with 2-H and not with C-11. The NOE experiment also confirmed the assignment of the 1H spectrum.
9,11-Dihydroindolizino[1,2-b]quinolin-9-one 29a48. A solution of (Z)-1-(2-bromo-3-phenylprop-2-enyl)-6-oxo-1,6-dihydropyridine-2-carbonitrile 13a (150 mg, 0.476 mmol), hexamethylditin (1.2 g, 3.66 mmol) and di-tert-butyl peroxide (0.75 cm3, 4.10 mmol) in tert-butylbenzene (6.5 cm3) in a Schlenk tube was subjected to the freeze–thaw method (6 times). The sealed tube was immersed in an oil bath at 120 °C surrounded by a safety shield in a fumehood for 2 days. The reaction mixture was cooled to ambient temperature and the reaction mixture evaporated under reduced pressure. Purification as detailed in the light irradiation procedure gave 29a (54 mg, 50%). The mp, IR, MS and NMR spectra were identical to those of authentic material.
The same procedure using 13b (170 mg, 0.476 mmol), hexamethylditin (0.343 g, 1.05 mmol) and di-tert-butyl peroxide (0.148 cm3, 0.81 mmol) for 24 h gave 29a (0.066 g, 60%).
O); m/z (CI) 199 (M+, 1%), 172 (10), 154 (34), 127 (33), 126 (27), 98 (82), 82 (55), 71 (61) and 55 (100).Diethyl 2-(cyanomethyl)propanedioate (2.00 g, 10.0 mmol) was added to a stirred suspension of sodium hydride (0.29 g, 12.0 mmol) in tetrahydrofuran (30 cm3) at 0 °C under nitrogen. After 30 min, (Z)-1-(2,3-dibromoprop-1-enyl)benzene 8a (2.74 g, 10.0 mmol) was added and the mixture stirred for a further 18 h at room temperature. Aqueous hydrochloric acid (2 M, 20 cm3) was added and the product was extracted with dichloromethane, dried and evaporated to dryness, giving an orange oil. Purification by column chromatography gave diethyl 2-(2-bromo-3-phenylprop-2-enyl)-2-(cyanomethyl)propanedioate 35a as a pale yellow oil (2.87 g, 73%) [Found (M + NH4)+, 411.0916. (C18H2079BrNO4
+ NH4) requires 411.0920]; νmax/cm−1 2984, 2939, 2255,
1737, 1446, 1369, 1297, 1211, 1093, 913, 734 and 696; δH (400 MHz) 1.29–1.32 (6 H, t, J 7.2, Me), 3.12 (2 H, s, CH2CN), 3.54 (2 H, s, CH2C), 4.22–4.35 (4 H, m, OCH2), 7.03 (1 H, s, vinyl-H), 7.24–7.39 (3 H, m, Ar–H) and 7.55 (2 H, dd, J 8.0 and 1.0, Ar–H); δC (100 MHz) 13.92 (Me), 21.47 (CH2CN), 45.19 (CH2), 55.09 (2-C), 62.86 (OCH2), 116.36 and 117.80 (CBr and CN), 128.22, 128.43 and 128.98 (Ph-CH), 135.00 (Ph 1-C) and 167.86 (CO); m/z (CI) 413 [(M + NH4)+, 68%], 411 (67), 287 (15), 217 (100), 178 (17) and 106 (18).
O) and 202.29 (CO); m/z 316 (M+, 26%), 243 (49), 242 (100), 213 (30), 141 (77), 115 (94), 77 (8) and 40 (33).Acknowledgements
We thank the EPSRC for postdoctoral Research Associate grants (M.O.C. and C.F.B.) and the EPSRC Mass Spectrometry Unit, Swansea University, Wales for mass spectra.References
- M. Potmesil and H. Pinedo, Camptothecins: New Anticancer Agents, CRC Press, Boca Raton, Florida, 1995 Search PubMed.
- Some leading references: M. Toyota, C. Komori and M. Ihara, J. Org. Chem., 2000, 65, 7110 Search PubMed; D. L. Boger and J. Hong, J. Am. Chem. Soc., 1998, 120, 1218 CrossRef CAS.
- Some leading references: R. Mekouar, Y. Génisson, S. Leue and A. E. Greene, J. Org. Chem., 2000, 65, 5212 Search PubMed; I. Pendrak, S. Barney, R. Wittrock, D. M. Lambert and W. D. Kingsbury, J. Org. Chem., 1994, 59, 2623 CrossRef.
- R. T. Brown, L. Jianli and C. A. M. Santos, Tetrahedron Lett., 2000, 41, 859 CrossRef CAS.
- (a) H. Josien, S.-B. Ko, D. Bom and D. P. Curran, Chem. Eur. J., 1998, 4, 67 CrossRef CAS; (b) B. Dom, D. P. Curran, S. Kruszewski, S. G. Zimmer, J. T. Strode, G. Kohlhagen, W. Du, A. J. Chavan, K. A. Fraley, A. L. Bingcang, L. J. Latus, Y. Pommier and T. G. Burke, J. Med. Chem., 2000, 43, 3970 CrossRef; (c) H. Josien, D. Bom, D. P. Curran, Y.-H. Zheng and T.-C. Chou, Bioorg. Med. Chem. Lett., 1997, 7, 3189 CrossRef CAS; (d) H. Liu and D. P. Curran, J. Am. Chem. Soc., 1992, 114, 5863 CrossRef CAS.
- D. P. Curran, H. Liu, H. Josien and S.-B. Ko, Tetrahedron, 1996, 52, 11385 CrossRef CAS; D. P. Curran, S.-B. Ko and H. Josien, Angew. Chem., Int. Ed. Engl., 1995, 34, 2683 CrossRef CAS.
- D. P. Curran and H. Liu, J. Am. Chem. Soc., 1991, 113, 2127 CrossRef CAS.
- H. Josien and D. P. Curran, Tetrahedron, 1997, 53, 8881 CrossRef CAS.
- M.-L. Bennasar, C. Juan and J. Bosch, Chem. Commun., 2000, 2459 RSC.
- W. R. Bowman, C. F. Bridge, M. O. Cloonan and D. C. Leach, Synlett, 2001, 765 CrossRef CAS.
- W. R. Bowman, M. O. Cloonan and S. L. Krintel, J. Chem. Soc., Perkin Trans. 1, 2001, 2885 RSC; W. R. Bowman, C. F. Bridge and P. Brookes, J. Chem. Soc., Perkin Trans. 1, 2000, 1 RSC; F. Aldabbagh and W. R. Bowman, Contemp. Org. Synth., 1997, 4, 261 RSC.
- M. Tokuyama, Y. Kaburagi, X. Chen and T. Fukuyama, Synthesis, 2000, 429 CrossRef CAS; J. D. Rainier, A. R. Kennedy and E. Chase, Tetrahedron Lett., 1999, 40, 6325 CrossRef CAS; J. A. Murphy, K. A. Scott, R. S. Sinclair, C. Gonzalez Martin, A. R. Kennedy and N. Lewis, J. Chem. Soc., Perkin Trans. 1, 2000, 2395 RSC; J. A. Murphy, K. A. Scott, R. S. Sinclair and N. Lewis, Tetrahedron Lett., 1997, 38, 7295 CrossRef CAS; T. Fukuyama, X. Chen and G. Peng, J. Am. Chem. Soc., 1994, 116, 3127 CrossRef CAS.
- M. T. Lyon, S. Lawrence, D. J. Williams and Y. A. Jackson, J. Chem. Soc., Perkin Trans. 1, 1999, 437 RSC; Y. A. Jackson, M. T. Lyon, N. Townsend, K. Bellabe and F. Soltanik, J. Chem. Soc., Perkin Trans. 1, 1999, 205 Search PubMed; E. Kashiyama, I. Hutchinson, M.-S. Chua, S. F. Stinson, L. R. Phillips, G. Kaur, E. A. Sausville, T. D. Bradshaw, A. D. Westwell and M. F. G. Stevens, J. Med. Chem., 1999, 42, 4172 CrossRef CAS; I. Hutchinson, M. F. G. Stevens and A. D. Westwell, Tetrahedron Lett., 2000, 41, 425 CrossRef CAS.
- D. C. Harrowven and M. I. T. Nunn, Tetrahedron Lett., 1998, 39, 5875 CrossRef CAS; D. C. Harrowven, M. I. T. Nunn, N. J. Blumire and D. R. Fenwick, Tetrahedron Lett., 2000, 41, 6681 CrossRef CAS.
- D. Melandri, P. C. Montevecchi and M. L. Navacchia, Tetrahedron, 1999, 55, 12227 CrossRef CAS; P. C. Montevecchi and M. L. Navacchia, Tetrahedron Lett., 1998, 39, 9077 CrossRef CAS; P. C. Montevecchi, M. L. Navacchia and P. Spagnolo, Tetrahedron, 1998, 54, 8207 CrossRef CAS; R. A. Aitken, G. Burns and J. J. Morrison, J. Chem. Soc., Perkin Trans. 1, 1998, 3937 RSC.
- D. Nanni, G. Calestani, R. Leardini and G. Zanardi, Eur. J. Org. Chem., 2000, 707 CrossRef CAS; D. Nanni, P. Pareschi, C. Rizzoli, P. Sgarabotto and A. Tundo, Tetrahedron, 1995, 51, 9045 CrossRef CAS; I. Lenoir and M. L. Smith, J. Chem. Soc., Perkin Trans. 1, 2000, 641 RSC; P. C. Montevecchi, M. L. Navacchia and P. Spagnolo, Tetrahedron, 1998, 54, 8207 CrossRef CAS.
- C. M. Camaggi, R. Leardini, D. Nanni and G. Zanardi, Tetrahedron, 1998, 54, 5587 CrossRef CAS.
- Leading references: P. Double and G. Pattenden, J. Chem. Soc., Perkin Trans. 1, 1998, 2005 Search PubMed; O. Callaghan, C. Lampard, A. R. Kennedy and J. A. Murphy, Tetrahedron Lett., 1999, 40, 2225 RSC; N. Edwards, J. A. Macritchie and P. J. Parsons, Tetrahedron Lett., 1998, 39, 3605 CrossRef CAS.
- W. R. Bowman, M. J. Broadhurst, D. R. Coghlan and K. A. Lewis, Tetrahedron Lett., 1997, 38, 6301 CrossRef CAS; W. R. Bowman, D. R. Coghlan and H. Shah, C. R. Acad. Sci., Ser. IIc: Chim., 2001, 4, 625 CrossRef CAS and references therein.
- W. R. Bowman, P. T. Stephenson and A. R. Young, Tetrahedron, 1996, 52, 11445 CrossRef CAS and references therein.
- Review: G. K. Friestad, Tetrahedron, 2001, 57, 5461 Search PubMed.
- Review: A. G. Fallis and I. M. Brinza, Tetrahedron, 1997, 53, 17543 Search PubMed.
- Review: S. Z. Zard, Synlett, 1996, 1148 Search PubMed.
- M.-H. Le Tadic-Biadatti, A.-C. Callier-Dublanchet, J. H. Horner, B. Quiclet-Sire, S. Z. Zard and M. Newcomb, J. Org. Chem., 1997, 62, 559 CrossRef CAS.
- W. R. Bowman, C. F. Bridge and P. Brookes, Tetrahedron Lett., 2000, 41, 8989 CrossRef CAS.
- D. Griller, P. Schmid and K. U. Ingold, Can. J. Chem., 1979, 57, 831 CAS.
- P. Gros and Y. Fort, J. Chem. Soc., Perkin Trans. 1, 1998, 3515 RSC.
- H. Liu, S.-B. Ko, H. Josien and D. P. Curran, Tetrahedron Lett., 1995, 36, 8917 CrossRef CAS.
- W. R. Bowman and C. F. Bridge, Synth. Commun., 1999, 29, 4051 CAS.
- I. Hagedorn and W. Hohler, Angew. Chem., Int. Ed. Engl., 1975, 14, 486 CrossRef.
- H. Keskin, R. E. Miller and F. F. Nord, J. Org. Chem., 1951, 16, 199 CrossRef CAS; L. H. Klemm and K. W. Gopinath, J. Heterocycl. Chem., 1965, 2, 225 Search PubMed.
- W. M. F Fabian, C. O. Kappe and V. A. Bakulev, J. Org. Chem., 2000, 65, 47 CrossRef CAS.
- D. H. Hey, G. H. Jones and M. J. Perkins, J. Chem. Soc., Chem. Commun., 1969, 1375 RSC; D. H. Hey, G. H. Jones and M. J. Perkins, J. Chem. Soc. (C), 1971, 116 RSC.
- D. Crich and J.-T. Hwang, J. Org. Chem., 1998, 63, 2765 CrossRef CAS.
- W. R. Bowman, E. Mann and J. Parr, J. Chem. Soc., Perkin Trans. 1, 2000, 2991 RSC; review: A. Studer and M. Bossart, in Radicals in Organic Synthesis, eds. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, 2001, p. 62 Search PubMed.
- F. Aldabbagh, W. R. Bowman, E. Mann and A. M. Z. Slawin, Tetrahedron, 1999, 55, 8111 CrossRef CAS.
- W. R. Bowman, H. Heaney and B. M. Jordan, Tetrahedron, 1991, 47, 10119 CrossRef CAS.
- J. A. Howard, J. C. Tait and S. B. Tong, Can. J. Chem., 1979, 57, 2761 CAS; I. P. Beletskaya, A. S. Sigeev, V. A. Kuzmin and A. S. Tatikolov, J. Chem. Soc., Perkin Trans. 2, 2000, 107 RSC.
- I. Omae, Organotin Chemistry, Elsevier, Amsterdam, 1989, p. 181 Search PubMed.
- M. Harendza, J. Junggebauer, K. Leßmann, W. P. Neumann and H. Tews, Synlett, 1993, 286 CrossRef CAS; S. Kim, I. Y. Lee, J.-Y. Yoon and D. H. Oh, J. Am. Chem. Soc., 1996, 118, 5138 CrossRef CAS.
- M. C. Wani, A. W. Nicholas and M. E. Wall, J. Med. Chem., 1986, 29, 235.
- D. C. Harrowven, M. I. T. Nunn, N. A. Newman and D. R. Fenwick, Tetrahedron Lett., 2001, 42, 961 CrossRef CAS.
- J. T. Braunholtz, K. B. Mallion and F. G. Mann, J. Chem. Soc., 1962, 4346 RSC.
- F. Aldabbagh and W. R. Bowman, Tetrahedron Lett., 1997, 38, 3793 CrossRef CAS; F. Aldabbagh and W. R. Bowman, Tetrahedron, 1999, 55, 4109 CrossRef CAS; F. Aldabbagh, W. R. Bowman and E. Mann, Tetrahedron Lett., 1997, 38, 7937 CrossRef CAS.
- R. Leardini, D. Nanni, P. Pareschi, A. Tundo and G. Zanardi, J. Org. Chem., 1997, 62, 8394 CrossRef CAS.
- Y. Tominaga, H. Okuda and H. Mazume, J. Heterocycl. Chem., 1991, 28, 1245 Search PubMed.
- F. Coulomb, M.-L. Roumestant and J. Gore, Bull. Soc. Chim. Fr., 1973, 3352 Search PubMed.
- T. Kametani, H. Nomoto, H. Takoda and S. Takano, Tetrahedron, 1970, 26, 5753 CrossRef CAS.
- T. Sakamoto, S. Kaneda, S. Nishimura and H. Yamanaka, Chem. Pharm. Bull., 1985, 2, 565 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: additional experimental details. See http://www.rsc.org/suppdata/p1/b1/b108323f/ |
| This journal is © The Royal Society of Chemistry 2002 |