Influence of preparation procedure on polymer composition: synthesis and characterisation of polymethacrylates bearing β-D-glucopyranoside and β-D-galactopyranoside residues
Moira Ambrosia, Andrei S. Batsanova, Neil R. Cameron*a, Benjamin G. Davis*b, Judith A. K. Howarda and Rob Hunterc
aDepartment of Chemistry, University of Durham, South Road, Durham , UK DH1 3LE
bDyson Perrins Laboratory, Department of Chemistry, South Parks Road, Oxford, UK OX1 3QY
cUnilever Research Port Sunlight, Quarry Road East, Bebington, Wirral, UK CH63 3JW
First published on 11th December 2001
Abstract
Methacrylate derivatives bearing β-D-glucopyranoside and β-D-galactopyranoside residues are synthesised by glycosylation of 2-hydroxyethyl methacrylate (HEMA) with 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide and 2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl bromide, respectively. β-Selectivity in the glycosylation reactions is ensured by neighbouring-group participation of acetyl groups at O-2 in the glycosyl donors. 2-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucosyloxy)ethyl methacrylate (AcGlcEMA, 1a) was obtained as a crystalline solid and its crystal structure was determined by single-crystal X-ray diffraction. Deprotected polymers are synthesised in two parallel ways; either polymerisation of the protected monomers and subsequent deacetylation of the resulting polymers, or polymerisation of the previously deprotected monomers. The number- and weight-average relative molecular masses of both the protected and deprotected polymers are determined by size exclusion chromatography (SEC). Absolute molecular masses are obtained using the previously estimated refractive-index increments, dn/dc. It is found that polymerisation of deprotected monomers leads to polymers of well-defined composition, in contrast to the deacetylation of protected polymers.
Introduction
Polymers consisting of a chemically and biologically stable C–C backbone and a hydrophilic saccharide moiety in the side chain are called ‘glycopolymers’.1 Owing to their application in basic biochemical and biomedical research such as molecular-recognition processes, drug-delivery systems, affinity chromatography and cell-culture systems, glycopolymers have attracted increasing attention since they were first developed by Horejsi et al. in 1978.2The synthesis of carbohydrate-based polymers usually requires the preparation of polymerisable sugar derivatives,3–22 although the less frequently employed glycosylation of polymers is also possible.23,24 Different types of glycopolymers have been synthesised, mainly polyacrylamides,9,12 polystyrenes,11,13–15 polyacrylates3,19–21 and polymethacrylates.3–6 Kobayashi et al.7 have synthesised a variety of carbohydrate-containing polystyrene derivatives, which have been used as cell-specific biomedical materials. In particular, lactose-carrying polystyrene (PVLA) has been shown to be a useful substratum for the culture of hepatocytes. The synthesis of another type of polystyrene derivative bearing lactose or N,N′-diacetylchitobiose residues and the investigation of their interaction with lectins by means of a two-dimensional immunodiffusion test in agar and inhibition of haemoagglutinating activity was reported by Kobayashi et al.11 It was found that the lectin-poly(p-vinylbenzamido)-β-diacetylchitobiose binding was increased 103-times compared with that of the oligosaccharide itself. The attachment of different kinds of cells to tissue culture polystyrene plates coated with polyacrylamide containing glucose residues has been investigated by Bahulekar et al.12 Ohno et al.13 reported the synthesis of PVLA obtained by means of a different route. The deprotected polymer, DODA-PVLA, was then used to prepare sugar-carrying liposomes, the galactose residues of which were specifically and effectively recognised by the galactose-specific lectin Ricinus communis agglutinin (RCA).
Due to the polyfunctionality of sugars, multistep reactions including protecting-group chemistry are typically required for their manipulation. Indeed, selective reactions of non-protected sugars to form polymerisable monomers have only been achieved by enzymatic catalysis.25,26 The removal of protecting groups can be carried out either before or after the polymerisation. When considering the point at which to deprotect, the lability of the monomer and, to a lesser extent, the polymer, together with the possibility of a non-quantitative deprotection of the polymer, all have to be taken in account. Usually, deprotection of the polymers is not quantitative; therefore the removal of protecting groups is, if possible, preferably carried out at the monomer stage.10 Nevertheless, to our knowledge, only two examples of deprotection at the monomer stage have been reported so far.6,9 Despite the importance of this strategy, no systematic comparison has been made between deprotection pre- or post-polymerisation.
In this paper we report the synthesis of methacrylate derivatives of glucose and galactose by glycosylation of 2-hydroxyethyl methacrylate (HEMA) following the procedure reported in Scheme 1. The protected monomers were polymerised with 2,2′-azoisobutyronitrile (AIBN) in chloroform at 65 °C and the resulting polymers, were subsequently deprotected with sodium methoxide in a 1 ∶ 1 mixture of,chloroform and methanol. Alternatively, the protected monomers were first deacetylated with sodium methoxide in methanol and then polymerised in a mixture 4 ∶ 1 of water and methanol at 65 °C using potassium persulfate as initiator.
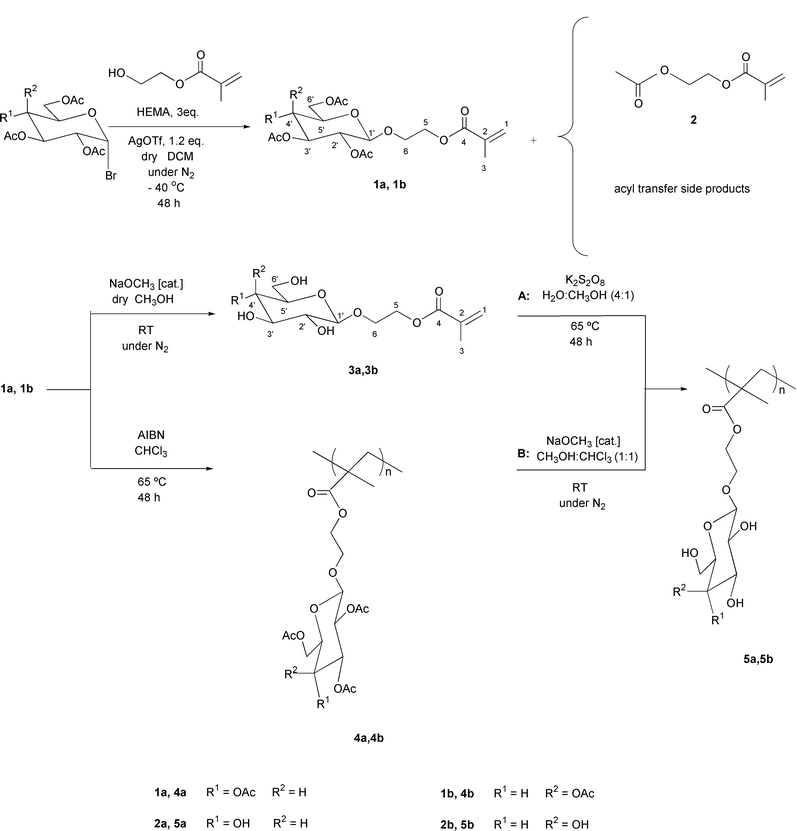 | ||
| Scheme 1 Monomer and polymer syntheses. | ||
Methacrylate and acrylate derivatives containing glucose and galactose have been synthesised previously. Kitazawa et al.3 reported the glycosylation of HEMA and 2-hydroxyethyl acrylate (HEA) using several methyl glycosides as glycosyl donors, including methyl glucoside and methyl galactoside, in the presence of phosphomolybdic acid as catalyst and 2,4-dinitrochlorobenzene as an inhibitor. However, the stereoselectivity of this method is low and an α,β anomeric mixture is obtained. Nakaya et al.5 synthesised 2-(2′,3′,4′,6′-tetra-O-acetyl-β-D-glucopyranosyloxy)ethyl methacrylate by reaction of HEMA with 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide by the method of Helferich,27 in the presence of silver oxide or mercury(II) cyanide, with yields of 54 and 58%, respectively. After free-radical polymerisation, the polymer obtained was deacetylated with sodium methoxide and the title polymer was identified by infrared spectroscopy data alone. 2-(2′,3′,4′,6′-Tetra-O-acetyl-β-D-glucopyranosyloxy)ethyl acrylate (AcGEA) was synthesised by Liang et al.19–21 according to the method of Helferich,27 by glycosylation of HEA with 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide using mercury(II) bromide as catalyst. After polymerisation by conventional free-radical and atom-transfer radical polymerisation, the polymer was deprotected with sodium-methoxide. The self-association tendency of poly(β-D-glucopyranosyloxy)ethyl acrylate (PGEA) in water was studied; a dependence of the critical aggregation concentration on the relative molecular mass of the polymer and on the temperature was observed. In order to understand the influence of hydrophobicity on the critical aggregation concentration, AcGEA was copolymerised with stearyl acrylate in different ratios. β-D-Galactopyranosyloxyethyl methacrylate has so far been synthesised exclusively by enzymatic catalysis.25,26
In light of these disparate routes, some with low yields or stereoselectivities and/or involving toxic mercury salts, we derived parallel routes to protected (1a,b) and deprotected (3a,b) carbohydrate glycoside esters. Not only was β-stereoselectivity in the glycosylation reactions ensured by neighbouring-group participation of acetyl groups at O-2 in the glycosyl donors, but also the yields of these monomer syntheses were considerably improved compared with previous syntheses. Moreover, all monomeric and polymeric products have been characterised fully by IR and NMR spectroscopy, optical activity measurements, elemental analysis and, for the monomers, mass spectrometry, leading to a more thorough characterisation than those reported so far3,5,19–21 – critical for a proper comparison of the influence of sugar type in biological applications. Absolute molecular masses were obtained for all polymers by size-exclusion chromatography (SEC) using the previously estimated refractive-index increments, dn/dc, determined using a calibrated light-scattering signal and working at known concentrations. Good characterisation, high purity, and a fully known composition are important requirements for the precise use and application of glycopolymers and for a true understanding of any results obtained. Therefore, in this article we report a detailed characterisation of the monomers and polymers and for the first time we describe how the polymer composition depends on the method of preparation used.
Results and discussion
1. Glycomonomers
The protected monomers AcGlcEMA 1a and AcGalEMA 1b were synthesised by coupling the glycosyl donors, 2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide and 2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl bromide, respectively, with the glycosyl acceptor 2-hydroxyethyl methacrylate (HEMA), in dry dichloromethane, using silver trifluoromethanesulfonate as catalyst, in good yields. The synthetic procedure is reported in Scheme 1. β-Stereoselectivity in the glycosylation reactions was ensured by neighbouring-group participation of acetyl groups at O-2 in the glycosyl donors. Acetyl transfer to the nucleophilic alcohol (HEMA) led to the formation of AcEMA 2 as the major side product. Such side reactions have been reported previously23,28–30 and may be attributed to rearrangements of orthoester intermediates.23 According to several proposals31,32 in the literature, the glycosylation reaction proceeds by activation of the glycosyl donor by silver trifluoromethanesulfonate, leading to the irreversible formation of a glycosyl oxocarbenium ion33 that, due to neighbouring-group participation, is in equilibrium with the corresponding carbocationic species.23,34 Nucleophilic attack of HEMA on the latter species can then result in the formation of the desired products 1a and 1b with AcEMA and monodeacetylated compounds as side products. Intramolecular neighbouring-group participation is expected to be kinetically favoured with respect to intermolecular nucleophilic attack so that the oxocarbenium ion is unlikely to have a long lifetime.35,36Products 1a, 1b, and 2 were isolated by flash column chromatography. 2-Acetoxyethyl methacrylate (AcEMA, 2) was characterised by NMR spectroscopy and mass spectrometry. In addition to 2, unchanged HEMA was also present in each crude product; this was not separable from the desired product by column chromatography. This problem was overcome by acetylation of HEMA at the end of the glycosylation reaction to give 2, which was removed easily by flash chromatography. In this manner, AcGlcEMA 1a and AcGalEMA 1b were obtained in yields of 72 and 80%, respectively – a significant advance on previously reported yields. They were characterised
by IR and NMR spectroscopy, optical activity measurements, mass spectrometry and elemental analysis. Both glycomonomers were identified by 1H-NMR spectroscopy as being the β-anomers. AcGlcEMA was obtained as a colourless crystalline solid and its crystal structure was determined by single-crystal X-ray diffraction (Fig. 1). The heterocycle adopts a normal chair conformation, with all the substituents in equatorial orientations. The olefinic C(10)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(12) bond and the adjacent ester group are nearly coplanar: the O(9)C(9)C(10)C(12) torsion angle is 3.1(3)°.
C(12) bond and the adjacent ester group are nearly coplanar: the O(9)C(9)C(10)C(12) torsion angle is 3.1(3)°.
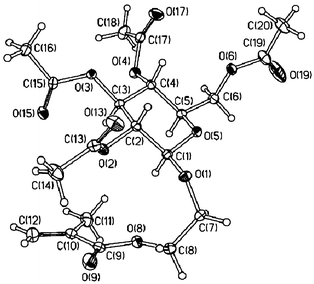 | ||
| Fig. 1 Crystal structure of 1a, showing 50% thermal ellipsoids. | ||
AcGalEMA 1b was obtained as colourless oil that was stored at 4 °C after addition of a radical-polymerisation inhibitor (hydroquinone, 10 ppm). AcGlcEMA and AcGalEMA are soluble in chloroform, methanol, dichloromethane, tetrahydrofuran, acetone, benzene and DMF.
The monomers were deprotected using a catalytic quantity of sodium methoxide in methanol, as reported in Scheme 1. The reaction, monitored continuously by TLC (acetonitrile–water, 9 ∶ 1), was stopped when the product resulting from the cleavage of the ester bond of the HEMA moiety was observed (as a spot having Rf = 0.2). The purification of GlcEMA 3a and GalEMA 3b by chromatography (chloroform–methanol, 8 ∶ 2) afforded the products in high purity. GlcEMA and GalEMA were obtained as strongly hygroscopic, amorphous, colourless solids, in yields of 80 and 75%, respectively. They were fully characterised for the first time by IR and NMR spectroscopy, optical activity measurements, mass spectrometry and elemental analysis. ES-mass spectra showed only one signal each, corresponding to the completely deprotected products. IR spectra showed the complete disappearance of the carbonyl absorption bands at ≈1750 cm−1 corresponding to the O-acetyl protecting groups. No signal due to the protecting groups could be seen in the 1H- and 13C-NMR spectra. Differences characteristic of acetyl removal in the 1H-NMR spectra were observed.
GlcEMA and GalEMA are soluble in methanol, DMF, water and, sparingly, in chloroform.
2. Glycopolymers
pAcGlcEMA 4a and pAcGalEMA 4b were obtained as white solids by polymerising 1a and 1b, respectively, with AIBN in chloroform at 65 °C for 48 h. The IR spectra showed that the vinyl absorption bands (1321 and 1299 cm−1, 1320 and 1295 cm−1, for AcGlcEMA and AcGalEMA, respectively) had disappeared. Moreover, no signals due to vinyl protons and carbons could be seen in the 1H- and 13C-NMR spectra. As for the corresponding monomers, pAcGlcEMA and pAcGalEMA showed optical activity due to the saccharide units (the specific rotations [α]TD, measured in chloroform, are reported in the Experimental section). pAcGlcEMA and pAcGalEMA are soluble in chloroform, THF, benzene, DMF and acetone.The number- and weight-average relative molecular masses were determined by size-exclusion chromatography (SEC). Absolute molecular mass values were obtained using the previously estimated refractive-index increments, dn/dc, which were determined in THF at 30 °C using a calibrated light-scattering signal, working at known concentrations and with a He–Ne laser (wavelength 670 nm) as the light source. Values of 0.058 ml g−1 and 0.071 ml g−1 for pAcGlcEMA and pAcGalEMA, respectively, were obtained. These values were consistent for three batches of the same polymer. Since the experimental conditions were the same for both polymer solutions, this interesting difference could possibly be explained by different conformations of the two polymers in solution perhaps induced by the different side-chain stereochemistries. The average relative molecular masses determined using these dn/dc values are reported in Table 1.
| Polymer | Mn/g mol−1 | Mw/g mol−1 | Mw/Mn | (dn/dc)/ml g−1 |
|---|---|---|---|---|
| pAcGlcEMA 4a | 61 600 | 135 900 | 2.21 | 0.058 ± 0.001 |
| pAcGalEMA 4b | 63 000 | 156 870 | 2.49 | 0.071 ± 0.002 |
| pGlcEMA 5aA | 23 000 | 60 000 | 2.61 | 0.131 |
| pGalEMA 5bA | 461 000 | 1 019 000 | 2.21 | 0.140 |
| pGlcEMA 5aB | 5 932 | 10 410 | 1.75 | 0.126 |
| pGalEMA 5bB | 2 984 | 6 410 | 2.15 | 0.146 |
The deprotected polymers, pGlcEMA 5a and pGalEMA 5b, were obtained following the two different procedures reported in Scheme 1. In route A, the deprotected monomers 3a and 3b were polymerised with potassium persulfate in a 4 ∶ 1 mixture of water and methanol at 65 °C for 48 h. In route B, the protected polymers 4a and 4b were deacetylated with sodium methoxide in a 1 ∶ 1 mixture of chloroform and methanol. In both cases, 5a and 5b were obtained as highly hygroscopic white solids. The two polymers synthesised by polymerisation of the deprotected monomers are denoted by (A); polymers obtained by deacetylating the protected polymers are denoted by (B). pGlcEMA and pGalEMA were characterised by IR and NMR spectroscopy, optical activity measurements and elemental analysis. All the four deprotected polymers synthesised were soluble only in water and the resulting aqueous solutions tended to foam on agitation, perhaps indicating surface activity. Measurements of surface tension and critical-aggregation concentration are in progress.
The 1H-NMR spectra of the polymers prepared by each route showed noticeable differences, as reported in Fig. 2 for pGalEMA(A) and pGalEMA(B). It can be seen that, despite exhaustive deprotecting conditions, signals due to methyl protons of the protecting acetyl groups can still be seen in the range δ 2.0–2.2 in the spectrum of pGalEMA(B), while no trace of these signals is present in the spectrum of pGalEMA(A). The presence in pGalEMA(B) of some residual protecting groups was also shown in the 13C-NMR spectrum by peaks in the range δC 22.0–24.0. Moreover, microanalyses of pGalEMA(B) typically gave a higher carbon percentage than expected (see the Experimental section). In only one case was the result in agreement with the calculated composition. The same results were found for pGlcEMA(A) and pGlcEMA(B). It should be noted that deprotection could not be continued to completion due to precipitation of the partially deprotected polymers from solution. Moreover, cleavage of the methacrylate ester bonds could occur during the deacetylation of pAcGlcEMA and pAcGalEMA, leading to polymers containing methyl ester and/or acid functionalities in the side chains. While there is no clear evidence to support this, the 1H-NMR spectrum of pGalEMA(B) contains many more weak signals just above the baseline, indicating more impurities than in pGalEMA(A) (Fig. 2). Some of these resonances are in regions where the OMe signals of methyl methacrylate units would be expected (δ ≈3.5).
 | ||
| Fig. 2 1H-NMR spectra of pGalEMA(A) and pGalEMA(B). | ||
Absolute relative molecular masses of the deprotected polymers were determined by SEC in aqueous solution and the obtained values are reported in Table 1. The refractive-index increments, dn/dc, were determined by means of a differential refractometer, working under the same experimental conditions as used for the SEC measurements. Values of 0.131 ml g−1, 0.140 ml g−1, 0.126 ml g−1 and 0.146 ml g−1 for pGlcEMA(A), pGalEMA(A), pGlcEMA(B) and pGalEMA(B), respectively, were obtained.
The average molar mass values of 5bA are much higher than those of 5aA, which may be indicative of a tendency of the former to aggregate more strongly in aqueous solution (it was noticed that aqueous solutions of the deprotected polymers had a tendency to froth). However, more surprising is the difference in average molar mass values of deprotected polymers prepared by route B compared with their parent polymers (compare 5aB with 4a and 5bB with 4b). While a reduction in relative molecular mass is expected due to removal of four acetyl groups per sugar residue, this alone cannot account for the differences observed in Mn and Mw between 5B and 4. Scission of polymer chain C–C bonds during deprotection is an unlikely explanation. The average molar mass values for 5B polymers may in fact be artefacts of the polymer-deprotection procedure. During the deprotection, a white polymeric precipitate is produced, which is collected as the product. Since the chains become less soluble in the reaction medium as the extent of deprotection of each chain increases, precipitation of shorter chains is likely to occur before that of longer chains. Therefore, we may have inadvertently fractionated our polymer samples during deprotection, leaving the longer chains in solution. This serves to highlight the differences between the glycopolymer preparation routes and reinforces our belief in the unreliability of the polymer deprotection route (B).
Experimental
General
2,3,4,6-Tetra-O-acetyl-α-D-glucopyranosyl bromide (>95%) and 2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl bromide ≈95%) were purchased from Sigma and Fluka, respectively. 2-Hydroxyethyl methacrylate (HEMA, >99%), silver trifluoromethanesulfonate (>99%), acetic anhydride (>99%), potassium persulfate (>99%), pyridine (99.8%), methanol (99.8%) and cation-exchange resin DOWEX × 50W × 2-200 were purchased from Aldrich. 2,2′-Azoisobutyronitrile (AIBN, 97%) was obtained from BDH Laboratory Supplies. HEMA was purified by distillation under vacuum; dichloromethane (DCM) was distilled from calcium hydride under N2; all other chemicals were used without further purification.NMR spectra were recorded with a Varian Inova 500 spectrometer, operating at 499.78 (1H) and at 125.67 MHz (13C). IR spectra were obtained with a Perkin-Elmer 1600 Series FTIR spectrometer. Mass spectra were obtained with a Micromass Platform spectrometer and with a Micromass LCT spectrometer, ionisation modes ES+ or ES−. Size-exclusion chromatography (SEC) of pAcGlcEMA and of pAcGalEMA was performed with a Viscotel 200 + light scattering, in THF, using a Plgel 10 μ MIXED-B column and working with a flow rate of 1.000 ml min−1 and an injection volume of 100 μl. Absolute molecular masses of pGlcEMA(A,B) and pGalEMA(A,B) were determined by aqueous size-exclusion chromatography coupled to a Water 410 RI detector and a Wyatt DAWN DSP MALLS, using a TSK GMPW column (30 cm × 7.8 mm id). The mobile phase consisted of 80% HPLC-grade water, 20% methanol, 0.05 M NaNO3 and 2.5 ml l−1 1.0 M NaOH at a flow rate of 0.8 ml min−1. The MALLS detectors were normalised using 50K pullulan purchased from Gearing Scientific. All polymer samples were prepared as 0.2–0.6 wt% solutions and filtered through 0.2 μm filters. Molecular mass averages and polydispersity were calculated using Astra 32 software. The dn/dc values used were previously determined using a model BP-2000-V Brice-Phoenix Visual Laboratory Type Differential Refractometer, Phoenix Precision Instrument Company, Pennsylvania (USA); four solutions of concentration ranging between ca. ≈4% and 15% in 20% methanol, 80% 0.05 M NaNO3 and 2.5 ml l−1 1.0 M NaOH solution were prepared for each polymer and the measurements carried out at 25 °C. Optical rotations were estimated using a P-1020 Series Polarimeter, JASCO (UK) Ltd. [α]D-Values are in units of 10−1 deg cm2 g−1. Elemental analyses were obtained with an Exeter Analytical Inc. CE-440 Elemental Analyzer. Products were freeze-dried using a Christ ALPHA-1-4 freeze-dryer with controller LDC-1M.
X-Ray crystallography†
The diffraction experiment (nominally covering the full sphere of reciprocal space) was carried out at T = 100 K on a SMART 3-circle diffractometer with a 1K CCD area detector,‡ using graphite-monochromated Mo-Kα radiation (λ = 0.710 73 Å) and a Cryostream (Oxford Cryosystems) open-flow N2 gas cryostat.Monomer and polymer syntheses
O of O-acetyl groups), 1718 (CO of HEMA moiety), 1635, 1321, 1299 (CC); δH¶ (500 MHz; CDCl3) 1.99 (3H, s, 3 × H-3), 2.00, 2.01, 2.03, 2.07 (3H × 4, 4s Ac × 4), 3.68 (1H, ddd, J4′,5′ 10.5 Hz, J5′,6a′ 2.5 Hz, J5′,6b′ 4.5 Hz, H-5′), 3.81 (1H, ddd, J6a,6b 11.5 Hz, J5a,6a 7.0 Hz, J5b,6a 3.5 Hz, H-6a), 4.04 (1H, ddd, J6a,6b 11.5 Hz, J5a,6b
=
J5b,6b
= 5.0 Hz, H-6b), 4.12 (1H, dd, J6a′,6b′
12.2 Hz, J5′,6a′ 2.5 Hz, H-6′a), 4.25 (1H, dd, J6a′,6b′ 12.2 Hz, J5′,6b′ 4.5 Hz, H-6′b), 4.23–4.32 (2H, m, 2 × H-5a,b), 4.56 (1H, d, J1′,2′ 7.9 Hz, H-1′), 4.99 (1H, dd, J1′,2′ 7.7 Hz, J2′,3′ 9.7 Hz, H-2′), 5.07 (1H, t, J 9.7 Hz, H-4′), 5.18 (1H, t, J 9.5 Hz, H-3′), 5.57–5.59 (1H, m, H-1 Z to CH3–CC), 6.10–6.12 (1H, m, H-1 E to CH3–CC); NOESY correlation studies allowed the differentiation between H-1(Z) and H-1(E) due to the long range coupling of H-1(Z) with 3 × H-3; NOESY also allowed the differentiation between 2 × H-5 and
2 × H-6 due to the long-range coupling of H-6 with H-1′; δC (125.67 MHz, decoupled 1H 500 MHz; CDCl3) 18.2 (C-3); 20.5, 20.6 (H3CCOO × 4), 61.8 (C-6′), 63.3 (C-5), 67.4 (C-6), 68.2 (C-4′), 71.0 (C-2′), 71.8 (C-5′), 72.6 (C-3′), 100.7 (C-1′), 125.8 (C-1), 135.9 (C-2), 167.1 (C-4), 169.2, 169.3, 170.2, 170.6 (H3CCOO × 4); results of HETCOR and COSY correlation studies have been used in order to assign the observed signals to the hydrogen and carbon atoms of the compound; LRMS m/z (ES+): Found: 483.5 (M + Na)+, 100%.O of acetate groups), 1724 (CO of HEMA
moiety), 1632, 1320, 1295 (CC); δH (500 MHz; CDCl3) 1.96 (3H, s, 3 × H-3), 1.99, 2.02, 2.03, 2.13 (3H × 4, 4 s, Ac × 4), 3.82 (1H, ddd, J6a,6b 11.5 Hz, J5a,6a 7.5 Hz, J5b,6a 4.0 Hz, H-6a), 3.90 (1H, td, Jt
=
J5′,6a′
=
J5′,6b′
= 6.5 Hz, Jd
=
J4′,5′
= 1.1 Hz, H-5′), 4.04 (1H, ddd, J6a,6b 11.5 Hz, J5a,6b
=
J5b,6b
= 5.0 Hz, H-6b), 4.10 (1H, dd, J6a′,6b′ 10.3 Hz, J5′,6a′ 6.7 Hz, H-6′a), 4.14 (1H, dd, J6a′,6b′ 10.9 Hz, J5′,6b′ 6.5 Hz, H-6′b), 4.26 (1H,
ddd, J5a,5b 12.0 Hz, J5a,6a 7.5 Hz, J5a,6b 4.5 Hz, H-5a), 4.31 (1H, ddd, J5a,5b 12.5 Hz, J5b,6a
=
J5b,6b
= 5.0 Hz, H-5b), 4.52 (1H, d, J1′,2′ 8.0 Hz, H-1′), 4.98 (1H, dd, J2′,3′ 10.5 Hz, J3′,4′ 3.5 Hz, H-3′), 5.19 (1H, dd, J1′,2′ 8.0 Hz, J2′,3′ 10.5 Hz, H-2′), 5.36 (1H, dd, J3′,4′ 3.5 Hz, J4′,5′ 1.0 Hz, H-4′), 5.57–5.58 (1H, m, H-1 Z to CH3–CC), 6.10–6.11 (1H, m, H-1 E to CH3–CC); δC (125.67 MHz, decoupled 1H 500 MHz; CDCl3) 18.2 (C-3),
20.5, 20.6 (H3CCOO × 4), 61.2 (C-6′), 63.4 (C-5), 66.9 (C-4′), 67.3 (C-6), 68.5 (C-2′), 70.6 (C-3′), 70.8 (C-5′), 101.2 (C-1′), 125.8 (C-1), 136.0 (C-2), 167.0 (C-4), 169.3, 170.1, 170.2, 170.3 (H3CCOO × 4); results of HETCOR and COSY correlation studies have been used in order to assign the observed signals to the hydrogen and carbon atoms of the compound; LRMS m/z (ES+): Found: 483.3 (M + Na)+, 100%.C), 2.05 (3H, s, CH3–CO), 4.26–4.36 (4H, m, CH2CH2), 5.56–5.58 (1H, m, vinyl proton Z to CH3–CC), 6.10 (1H, m, vinyl proton E to CH3–CC); LRMS m/z (ES+): Found: 195 (M + Na)+, 100%, 211 (M + K)+, 10.O of HEMA moiety), 1635, 1320, 1298 (CC); δH (500 MHz; CD3OD) 1.93 (3H, m, 3 × H-3), 3.17 (1H, dd, J1′,2′ 8.0 Hz, J2′,3′ 9.5 Hz, H-2′), 3.25–3.35 (3H, m, H-3′, H-4′, H-5′), 3.65 (1H, dd, J6a′,6b′ 12.0 Hz, J5′,6a′
5.5 Hz, H-6′a), 3.83 (1H, ddd, J6a,6b 11.5 Hz, J5a,6a 6.0 Hz, J5b,6a 3.5 Hz, H-6a), 3.84 (1H, dd, J6a′,6b′ 11.7 Hz, J5′,6b′ 1.7 Hz, H-6′b), 4.09 (1H, ddd, J6a,6b 11.5 Hz, J5a,6b 4.0 Hz, J5b,6b 6.0 Hz, H-6b), 4.29 (1H, ddd, J5a,5b 12.0 Hz, J5a,6a 5.5 Hz, J5a,6b 3.5 Hz, H-5a), 4.30 (1H, d, J1′,2′ 8.0 Hz, H-1′), 4.34 (1H, ddd, J5a,5b 12.0 Hz, J5b,6a 3.5 Hz, J5b,6b 6.0 Hz, H-5b), 5.61–5.62 (1H, m, H-1 Z to CH3–CC), 6.11–6.12 (1H, m, H-1 E to CH3–CC); δC (125.67 MHz, decoupled 1H
500 MHz; CD3OD) 18.4 (C-3), 62.7 (C-6′), 65.3 (C-5), 68.6 (C-6), 71.6 (C-3′ or C-4′ or C-5′), 75.0 (C-2′), 78.0 (2C, C-3′ or C-4′ or C-5′), 104.6 (C-1′), 126.4 (C-1), 137.6 (C-2), 168.9 (C-4) [HRMS m/z (ES+): Found: 315.1049 (M + Na)+. C12H20NaO8 requires m/z, 315.1056].O of HEMA moiety), 1636, 1320, 1298 (CC); δH (500 MHz; CD3OD) 1.93 (3H, m, 3 ×
H-3), 3.45 (1H, dd, J2′,3′ 9.7 Hz, J3′,4′ 3.3 Hz, H-3′), 3.50 (1H, td, Jt
=
J5′,6a′
=
J5′,6b′
= 5.5 Hz, Jd
=
J4′,5′
= 1.0 Hz, H-5′), 3.52 (1H, dd, J1′,2′ 7.5 Hz, J2′,3′ 10.0 Hz, H-2′), 3.71 (1H, dd, J6a′,6b′ 11.0 Hz, J5′,6a′ 5.5 Hz, H-6′a), 3.74 (1H, dd, J6a′,6b′ 11.0 Hz, J5′,6b′ 7.0 Hz, H-6′b), 3.82 (1H, dd, J3′,4′ 3.3 Hz, J4′,5′ 0.7 Hz, H-4′), 3.84 (1H, ddd, J6a,6b 12.0 Hz, J5a,6a
6.0 Hz, J5b,6a 4.0 Hz, H-6a), 4.10 (1H, ddd, J6a,6b 12.0 Hz, J5a,6b 3.7 Hz, J5b,6b 6.0 Hz, H-6b), 4.26 (1H, d, J1′,2′ 7.5 Hz, H-1′), 4.31 (1H, ddd, J5a,5b 12.0 Hz, J5a,6a 6.0 Hz, J5a,6b 3.5 Hz, H-5a), 4.35 (1H, ddd, J5a,5b 12.0 Hz, J5b,6a 3.5 Hz, J5b,6b 6.0 Hz, H-5b), 5.61–5.62 (1H, m, H-1 Z to CH3–CC), 6.12 (1H, m, H-1 E to CH3–CC); δC (125.67 MHz; decoupled 1H 500 MHz; CD3OD) 18.4 (C-3), 62.5 (C-6′), 65.3 (C-5), 68.5 (C-6), 70.3 (C-4′), 72.4 (C-2′), 74.9 (C-3′), 76.7 (C-5′), 105.3 (C-1′),
126.4 (C-1), 137.7 (C-2), 168.8 (C-4) [HRMS m/z (ES+): Found: 315.1065 (M + Na)+. C12H20NaO8 requires m/z, 315.1056].O of acetate groups); δH (500 MHz; CDCl3) 0.75–1.15 (3H, br m, CH3–C), 1.60–2.27 (2H, br, CH2), 2.01, 2.03, 2.06, 2.10 (12H, 4 s, Ac × 4), 3.70–3.88, 3.95–4.07, 4.10–4.25, 4.99–5.03, 5.08–5.18, 5.20–5.28 (10H, protons of the carbohydrate residue and the methylene groups of the side chains), 4.66 (1H, br, anomeric proton); δC (125.67 MHz, decoupled 1H 500 MHz; CDCl3) 6.5, 18.2 (1C, br, CH3–C, racemic–racemic (rr) and meso–racemic (mr) triads, respectively), 20.6, 20.7, 20.8 (4C, H3CCOO × 4),
45.2 (1C, CH2), 52.2–55.6 (1C, br, CH3–C), 61.1 (1C, carbohydrate residue of the side chains), 63.6 (1C, CH2O-carbohydrate residue), 66.6 (1C, CH2OCO), 67.3, 69.1, 70.6, 70.8 (4C, carbohydrate residue of the side chain), 100.5 (1C, anomeric carbon), 169.3, 170.0, 170.3 (4C, H3CCOO × 4), 176.5, 176.9–177.8 (1C, br, CCOO, meso–racemic (mr) and racemic–racemic (rr) triads, respectively); results of HETCOR correlation studies have been used in order to assign the observed signals to hydrogen and carbon atoms of the compound.O of acetate groups); δH (500 MHz; CDCl3) 0.78–1.16 (3H, br m, CH3–C), 1.63–2.20
(2H, br, CH2), 1.98, 2.05, 2.06, 2.16 (12H, 4 s, Ac × 4), 3.71–3.83, 3.95–4.09, 4.10–4.24, 5.05–5.21, 5.37–5.43 (10H, protons of the carbohydrate residue and the methylene groups of the side chains), 4.62 (1H, br, anomeric proton); δC (125.67 MHz; decoupled 1H 500 MHz; CDCl3) 17.4, 18.9 (1C, br, CH3–C, racemic–racemic (rr) and meso–racemic (mr) triads, respectively), 20.8, 20.9, 21.0 (4C, H3CCOO × 4), 45.2 (1C, CH2), 52.5–56.1 (1C, br, CH3–C), 61.3 (1C, carbohydrate residue of the side chains), 63.6 (1C, CH2O-carbohydrate residue), 66.6 (1C, CH2OCO), 67.3, 68.9, 70.9, 71.0 (4C, carbohydrate residue of the side chain), 101.1 (1C, anomeric carbon), 169.5, 170.2, 170.5 (4C, H3CCOO × 4), 176.5, 177.1–177.8
(1C, br, CCOO, meso–racemic (mr) and racemic–racemic (rr) triads, respectively); results of HETCOR correlation studies have been used in order to assign the observed signals to hydrogen and carbon atoms of the compound.Synthesis A. A solution of 3a (1 g, 3.4 mmol) and K2S2O8 (25 mg, 2.5 wt%) in a mixture of high-purity water–methanol (4 ∶ 1) was degassed by bubbling N2 through for 15 min at room temperature. Then, the flask was sealed and the polymerisation was carried out at 65 °C for 48 h. The resulting solution was freeze-dried and the recovered polymer was then purified by dialysis against water (Dialysis Tubing-Visking, Size 20 Inf. Dia. 18/32″ (14.3 mm): 30 M, MWCO–12–14 000 Daltons) for 1 week. The solution was freeze-dried to afford 5a (0.82 g, 82%) as a white hygroscopic solid; [α]22D −14.8 (c 0.12 in water); {Found: C, 48.82; H, 6.92. [(C12H20O8)m + 0.1 mol H2O] requires C, 49.01; H, 6.92%}; IR (KBr disc) ν/cm−1 3446 (OH), 1718 (C
O of HEMA moiety); δH (500 MHz; D2O) 0.73–1.22 (3H, br m, CH3–C), 1.78–2.22 (2H, br, CH2), 3.25–3.53, 3.67–3.76, 3.86–398, 4.08–4.32 (10H, protons of the carbohydrate residue and of the methylene groups of the side chains), 4.47 (1H, d, J 7.5 Hz, anomeric proton); δC (125.67 MHz; decoupled 1H 500 MHz; D2O) 17.2, 18.8, 21.2 (1C, br, CH3–C, racemic–racemic (rr), meso–racemic (mr) and meso–meso (mm) triads, respectively), 45.0 (1C, CH2), 51.0–53.7
(1C, br, CH3–C), 61.1 (1C, carbohydrate residue of the side chains), 65.1 (1C, CH2O-carbohydrate residue), 67.3 (1C, CH2OCO), 69.9, 73.3, 76.0, 76.1 (4C, carbohydrate residue of the side chain), 102.7 (1C, anomeric carbon), 178.9, 179.7–180.0 (1C, br, CCOO, meso–racemic (mr) and racemic–racemic (rr) triads, respectively).Synthesis B. A solution of 4a (0.20 g) in CHCl3–CH3OH (1 ∶ 1, 8 ml) was stirred at room temperature under N2 for 15 min. Then, 1 ml of a freshly prepared 1 M solution of sodium methoxide in methanol was added and the formation of a white precipitate was observed after around 30 s. After stirring at room temperature under N2 for 1 h, the solid was filtered off, dissolved in water, and a cation-exchange resin (DOWEX 50W × 2-200) was added in order to remove Na+ cations. The solution was stirred for 15 min before filtration to remove the resin. After purification by dialysis against water, the solution was freeze-dried to afford 5a (0.80 g, 63%) as a white hygroscopic solid [Found: C, 51.26; H, 6.69. (C12H20O8)m requires C, 49.31; H, 6.90%. Assuming that two O-acetyl groups were still present after the deprotection: (C16H24O10)m would require C, 51.06; H, 6.43%.]; IR (KBr disc) ν/cm−1 3423 (OH), 1718 (C
O of HEMA moiety); δH (500 MHz; D2O) 0.75–1.18 (3H, br m, CH3–C), 1.72–2.22 (2H, br, CH2), 2.06, 2.17, 2.20 (methyl protons of O-acetyl groups still present after the deprotection), 3.31–3.52, 3.72, 3.92, 4.05–4.36 (10H, protons of the carbohydrate residue and of the methylene groups of the side chains), 4.48 (1H, d, J 7 Hz, anomeric proton); δC (125.67 MHz; decoupled 1H 500 MHz; D2O) 17.3, 18.9 (1C, br, CH3–C, racemic–racemic (rr) and meso–racemic (mr) triads, respectively), 45.0 (1C,
CH2), 51.0–53.8 (1C, br, CH3-C), 61.1 (1C, carbohydrate residue of the side chains), 65.1 (1C, CH2O-carbohydrate residue), 67.3 (1C, CH2OCO), 69.9, 73.3, 76.0, 76.1 (4C, carbohydrate residue of the side chain), 102.7 (1C, anomeric carbon), 178.8, 179.8–180.0 (1C, br, CCOO, meso–racemic (mr) and racemic–racemic (rr) triads, respectively).Synthesis A. 3b (1 g, 3.4 mmol) was polymerised and the resulting polymer was purified according to the procedure described in the synthesis of 5a to afford 5b (0.86 g, 86%) as a white hygroscopic solid; [α]22D +8.0 (c 0.10 in water) {Found: C, 48.82; H, 6.92. [(C12H20O8)m + 0.1 mol H2O] requires C, 49.03; H 6.91%}; IR (KBr disc) ν/cm−1 3447 (OH), 1718 (C
O of HEMA moiety); δH (500 MHz; D2O) 0.82–1.20 (3H, br m, CH3–C),
1.78–2.35 (2H, br, CH2), 3.52–3.59, 3.62–3.72, 3.72–3.85, 3.89–4.02 4.06–4.35 (10H, protons of the carbohydrate residue and of the methylene groups of the side chains), 4.43 (1H, d, J 6.5 Hz, anomeric proton); δC (125.67 MHz; decoupled 1H 500 MHz; D2O) 17.2, 18.9 (1C, br, CH3–C, racemic–racemic (rr) and meso–racemic (mr) triads, respectively), 45.0 (1C, CH2), 52.0–53.8 (1C, br, CH3C), 61.1 (1C, carbohydrate residue of the side chains), 65.0 (1C, CH2O-carbohydrate residue), 67.1 (1C, CH2OCO), 68.8, 70.9, 73.0, 75.3 (4C, carbohydrate residue of the side chain), 103.2 (1C, anomeric carbon), 178.9, 179.8–180.0 (1C, br, CCOO, meso–racemic (mr) and racemic–racemic (rr) triads, respectively), 182.1 (COOH due to cleavage of the ester
bond of the side chains).Synthesis B. 4b (0.20 g) was deprotected and the resulting polymer purified as for 4a to afford 5b (0.83 g, 65%) as a white hygroscopic solid [Found: C, 51.20; H, 6.75. (C12H20O8)m requires C, 49.31; H, 6.90%. Assuming that two O-acetyl groups were still present after the deprotection (C16H24O10)m would require C, 51.06; H, 6.43%. IR (KBr disc) ν/cm−1 3445 (OH), 1718 (C
O of HEMA moiety); δH (500 MHz; D2O) 0.76–1.16 (3H, br m, CH3–C), 1.76–2.24 (2H, br, CH2), 2.07, 2.14, 2.20 (methyl protons of O-acetyl groups still present after the deprotection), 3.56, 3.61–4.00,
4.12–4.32 (10H, protons of the carbohydrate residue and of the methylene groups of the side chains), 4.43 (1H, d, J 6.5 Hz, anomeric proton); δC (125.67 MHz; decoupled 1H 500 MHz; D2O) 17.2, 18.8, 20.3 (1C, br, CH3–C, racemic–racemic (rr), meso–racemic (mr) and meso–meso (mm) triads, respectively), 23.8 (methyl carbon of O-acetyl groups still present after the deprotection), 45.0 (1C, CH2), 51.4–53.7 (1C, br, CH3–C), 61.1 (1C, carbohydrate residue of the side chains), 65.1 (1C, CH2O-carbohydrate residue), 67.1 (1C, CH2–OCO), 68.7, 70.9, 73.0, 75.3 (4C, carbohydrate residue of the side chain), 103.2 (1C, anomeric carbon), 178.9, 179.7–180.0 (1C, br, CCOO, meso–racemic (mr) and racemic–racemic (rr) triads,
respectively).Conclusions
Monomeric and polymeric methacrylate derivatives bearing β-D-glucopyranoside and β-D-galactopyranoside residues have been successfully synthesised in high yield and in an efficient stereocontrolled manner and have been fully characterised. Critically, it has been shown that deprotection of the polymers according to the methods used for all previous syntheses of poly(glyco-acrylates) and -methacrylates5,19–21 results in an incomplete deacetylation and yields products of ill-defined composition. Instead, fully deacetylated, well-defined and pure materials could be obtained by the novel method of polymerisation of the deprotected monomers. Absolute number- and weight-average relative molecular masses have been determined for all the polymers synthesised.A thorough comparison of all functional properties of these glycopolymers, including binding of the deprotected glycopolymers to specific receptor proteins (lectins) to assess their feasibility for the targeted delivery of bioactive species, is underway. Differences between the fully deprotected polymers obtained by route A and the partially acetylated polymers obtained by route B are expected.
Acknowledgements
We would like to thank Mr Doug Carswell (University of Durham) for performing SEC analyses in THF. We gratefully acknowledge the support of the EPSRC and the IRC in Polymer Science and Technology for the studentship to M. A. and the Royal Society for a Research Grant (no. 20408) to N. R. C.References
- T. Miyata and K. Nakamae, Trends Polym. Sci., 1997, 5, 198 Search PubMed.
- V. Horejsi, P. Smolek and J. Kocourek, Biochim. Biophys. Acta, 1978, 538, 293 CrossRef CAS.
- S. Kitazawa, M. Okumura, K. Kinomura and T. Sakakibara, Chem. Lett., 1990, 1733 CAS.
- K. Kobayashi, N. Kakishita, M. Okada, T. Akaike and S. Kitazawa, Makromol. Chem., Rapid Commun., 1993, 14, 293 Search PubMed.
- T. Nakaya, K. Nishio, M. Memita and M. Imoto, Makromol. Chem., Rapid Commun., 1993, 14, 77 Search PubMed.
- T. Nakaya, M. Memita and M. Kang, J. Macromol. Sci., Pure Appl. Chem., 1993, A30(suppl. 5), 349 Search PubMed.
- K. Kobayashi, A. Kobayashi, S. Tobe and T. Akaike, in Neoglycoconjugates: Preparation and Applications, Academic Press, Y. C. Lee and R. T. Lee (eds.), Academic Press, London, 1994, pp. 262–282 Search PubMed.
- G. Wulff and G. Clarkson, Macromol. Chem. Phys., 1994, 195, 2603 CrossRef CAS.
- T. Furuike, N. Nishi, S. Tokura and S.-I. Nishimura, Chem. Lett., 1995, 823 CAS.
- G. Wulff, J. Schmid and T. Venhoff, Macromol. Chem. Phys., 1996, 197, 259 CrossRef CAS.
- K. Kobayashi, A. Tsuchida, T. Usui and T. Akaike, Macromolecules, 1997, 30, 2016 CrossRef CAS.
- R. Bahulekar, T. Tokiwa, J. Kano, T. Matsumura, I. Kojima and M. Kodama, Carbohydr. Polym., 1998, 37, 71 CrossRef CAS.
- K. Ohno, T. Fukuda and H. Kitano, Macromol. Chem. Phys., 1998, 199, 2193 CrossRef CAS.
- K. Ohno, Y. Tsujii, T. Miyamoto, T. Fukuda, M. Goto, K. Kobayashi and T. Akaike, Macromolecules, 1998, 31, 1064 CrossRef CAS.
- K. Ohno, Y. Tsujii and T. Fukuda, J. Polym. Sci. Part A: Polym. Chem., 1998, 36, 2473 Search PubMed.
- K. Ohno, Y. Izu, S. Yamamoto, T. Miyamoto and T. Fukuda, Macromol. Chem. Phys., 1999, 200, 1619 CrossRef CAS.
- G. Wulff, H. Schmid and L. Zhu, Macromol. Chem. Phys., 1999, 200, 774 CrossRef CAS.
- X.-C. Liu and J. S. Dordick, J. Polym. Sci. Part A: Polym. Chem., 1999, 37, 1665 Search PubMed.
- Y.-Z. Liang, Z.-C. Li, G.-Q. Chen and F.-M. Li, Polym. Int., 1999, 48, 739 CrossRef CAS.
- Y.-Z. Liang, Z.-C. Li and F.-M. Li, J. Colloid Interface Sci., 2000, 224, 84 CrossRef CAS.
- Z.-C. Li, Y.-Z. Liang, G.-Q. Chen and F.-M. Li, Macromol. Rapid Commun., 2000, 21, 375 CrossRef CAS.
- H. Gruber and S. Knaus, Macromol. Symp., 2000, 152, 95 Search PubMed.
- T. Nukada, A. Berces, M. Z. Zgierski and D. M. Whitfield, J. Am. Chem. Soc., 1998, 120, 13291 CrossRef CAS.
- A. Takasu, T. Niwa, H. Itou, Y. Inai and T. Hirabayashi, Macromol. Rapid Commun., 2000, 21, 764 CrossRef CAS.
- S. Matsumura, H. Kubokawa and K. Toshima, Makromol. Chem., Rapid Commun., 1993, 14, 55 Search PubMed.
- T. Mori, S. Fujita and Y. Okahata, Carbohydr. Res., 1997, 298, 65 CrossRef CAS.
- B. Helferich and K. Weis, Chem. Ber., 1956, 89, 314 Search PubMed.
- R. U. Lemieux and A. R. Morgan, Can. J. Chem., 1965, 43, 2190 CAS.
- R. T. Lee and Y. C. Lee, Carbohydr. Res., 1995, 271, 131 CrossRef CAS.
- M.-Z. Liu, H.-N. Fan, Z.-W. Guo and Y.-Z. Hui, Carbohydr. Res., 1996, 290, 233 CrossRef CAS.
- R. U. Lemieux, Chem. Can., 1964, 16, 14 Search PubMed.
- G. Wulff and G. Röhle, Angew. Chem., Int. Ed. Engl., 1974, 13, 157 CrossRef CAS.
- H. K. Kondo, S. Aoki, Y. Ichikawa, R. L. Halcomb, H. Ritzen and C. H. Wong, J. Org. Chem., 1994, 59, 864 CrossRef CAS.
- N. K. Kochetkov, V. M. Zhulin, E. M. Klimov, N. N. Malysheva, Z. G. Makarova and A. Y. Ott, Carbohydr. Res., 1987, 164, 241 CrossRef CAS.
- N. S. Banait and W. P. Jencks, J. Am. Chem. Soc., 1991, 113, 7951 CrossRef CAS.
- C. Chiappe, G. Lo Moro and P. Munforte, Tetrahedron, 1997, 53, 10471 CrossRef CAS.
Footnotes |
| † CCDC reference number 171347. See http://www.rsc.org/suppdata/p1/b1/b108421f/ for crystallographic files in .cif or other electronic format. |
| ‡ SMART and SAINT, Area Detector Control and Integration Software. Version 6.0. Bruker AXS, Madison, Wisconsin, USA, 1999. |
| § SHELXTL, An Integrated System for Solving, Refining and Displaying Crystal Structures from Diffraction Data, Version 5.10. Bruker AXS, Madison, Wisconsin, USA, 1997. |
| ¶ See Scheme 1 for numbering scheme. |
| This journal is © The Royal Society of Chemistry 2002 |