4-Phenyloxazolidin-2-ones and isoindolin-1-ones: chiral auxiliaries for Diels–Alder reactions of N-substituted 1,3-dienes
Received
(in Cambridge, UK)
5th September 2001
, Accepted 14th November 2001
First published on 6th December 2001
Abstract
Terminally N-substituted dienes derived from 3-methylisoindolin-1-one, 4-isopropyl- and 4-phenyloxazolidin-2-one undergo Diels–Alder reaction with a range of activated dienophiles. The reactions reported are completely regio- and endo-selective, with the diastereoisomeric excess with respect to the auxiliary good to excellent in most of the cases reported. A model has been developed for rationalising the stereochemical outcome of these reactions.
Introduction
The chemistry of hetero-substituted dienes is rich, varied and has recently been reviewed.1 In particular, dienamides have emerged as reactive electron-rich dienes which readily participate in Diels–Alder reactions with electron-deficient dienophiles. Many of the pioneering studies on this class of compounds were carried out by the groups of Oppolzer2–4 and Overman.5–7 It was soon demonstrated that Diels–Alder reaction of dienamides with electron-deficient alkenes was completely regioselective, and gave only the isomer with 1,2-substitution of the electron-withdrawing group and nitrogen substituent.8,9 These reactions were also highly endo-selective. The first asymmetric Diels–Alder reaction utilising a dienamide employed a chiral dienophile10 and recently, chiral sulfoxides
have been incorporated as auxiliaries into N-dienes.11 Zezza and Smith reported the first Diels–Alder reactions of dienamides derived from lactams12 and this chemistry was extended to chiral dienamides derived from pyroglutamic acid.13 Although the asymmetric induction in the cycloaddition was excellent there was no possibility of removing the lactam whilst retaining the nitrogen functionality. However, if nitroso dienophiles were employed then the chiral pyroglutamate was readily removed after cycloaddition.14,15 The recent development of both chiral and achiral, highly reactive oxygenated dienamines and dienamides is showing great promise for the synthesis of cyclohexenes with densely packed functionality.16–22 Recently, catalytic asymmetric Diels–Alder reactions
of dienamides have been developed using chiral transition metal based Lewis acid catalysts23,24 and monoclonal antibodies.25 We previously communicated that chiral dienamides derived from 4-phenyloxazolidin-2-one and 3-methylisoindolin-1-one underwent Diels–Alder reactions with highly activated dienophiles and gave adducts with good to excellent de values,26 and now report these findings in detail.
Results and discussion
Since the Diels–Alder reaction of N-dienes is usually completely regio- and endo-selective, implying a highly ordered transition state, all that needs to be done to make the reaction enantioselective is to obtain face selectivity on either the diene or dienophile. Chiral dienamides were chosen as likely candidates to meet the above criteria and the first dienamide was prepared as outlined in Scheme 1. Hence, condensation of (S)-(−)-α-methylbenzylamine with crotonaldehyde gave an imine which on reaction with trimethylacetyl chloride and triethylamine in boiling benzene gave chiral dienamide 1 in 82% overall yield. Reaction of diene 1 with N-phenylmaleimide in boiling chloroform gave two diastereoisomeric products 2 and 3 (ratio
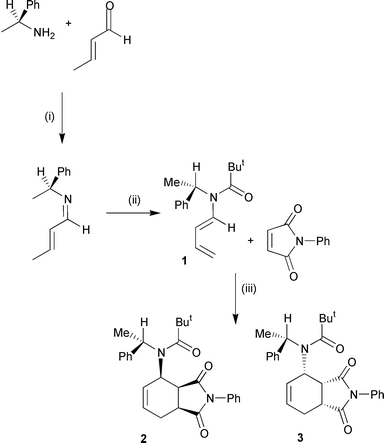
1 ∶ 2.5 by proton NMR spectroscopy) in 77% combined yield. These adducts were readily separated by flash chromatography. It was clear from the proton NMR spectra of compounds 2 and 3 that both had the same relative stereochemistry in the cyclohexene ring. In particular, J3a-H,4-H for isomers 2 and 3 was very similar at 9.5 and 9.8 Hz respectively. Although these values appear to be a little large for an axial–equatorial vicinal coupling constant, X-ray structural analysis of the highly crystalline minor adduct 2 (Fig. 1) confirmed that this was indeed an endo-isomer and the absolute stereochemistry was determined as (3a′S,4′R,7a′S,1″S).26Fig. 1 also clearly shows that the benzylic hydrogen atom of the auxiliary is in the same plane as the amide carbonyl group and is eclipsed by the tert-butyl group. This conformation is predicted to be of low energy as it minimises pseudo-1,3-allylic strain.27 It was taken that the major product was the alternative endo-isomer resulting from attack on the opposite face of the diene. The stereochemical outcome of this reaction can be rationalised as follows.
 |
| | Fig. 1 X-Ray structure of minor diastereoisomer 2. | |
(i) The diene adopted a conformation in which the amide carbonyl group was anti to the diene.
(ii) The auxiliary adopted a conformation in which the benzylic hydrogen was coplanar with the amide carbonyl group to minimise pseudo-allylic 1,3-strain.
(iii) N-Phenylmaleimide approached the diene from the face shielded by the smaller of the two remaining substituents on the auxiliary, i.e. from the face shielded by the methyl group.
If this empirical predictive model has any physical significance, then in order to improve the diastereoselectivity on cycloaddition it is necessary to have substituents attached to the chiral centre on the auxiliary which are sterically either more demanding than a phenyl group, or less demanding than a methyl group. Clearly, it is not a trivial task to design these features into an auxiliary. An alternative solution to this problem is to have the auxiliary and amide carbonyl group both present in a ring. This would ensure near coplanarity between the amide carbonyl group and the alkyl tether, leaving the possibility of having a hydrogen atom available for attachment to the auxiliary for control of face selectivity. This reasoning led to benzylic lactams and phenyloxazolidinones as second generation auxiliaries.
(S)-(+)-3-Methylisoindolin-1-one 4 was readily prepared by the literature procedure using (S)-(+)-N-(2,2-dimethylpropionyl)-1-phenylethylamine as starting material. However, the yield for the ortho-lithiation step was poor at 24% (Scheme 2).28 Chiral lactam 4 condensed with crotonaldehyde with azeotropic removal of water and gave the fairly sensitive diene 5 in 62% yield. The stereochemistry of the 1′,2′-alkene was exclusively E and this was readily assigned from the value of the coupling constant J1′-H,2′-H of 14.7 Hz. Diene 5 was more reactive than diene 1 and underwent Diels–Alder reaction with N-phenylmaleimide
to give endo-cycloadducts 6 and 7. The effect of temperature on the diastereoselectivity of this reaction is annotated in Table 1.
 |
| | Scheme 2
Reagents and conditions: ButLi, THF, −78 °C, then diethyl carbonate; (ii) crotonaldehyde, PPTS, benzene, 80 °C; (iii) chloroform, 4 °C, one week; (iv) 5 M LiClO4 in Et2O, 35 °C, 4 days. | |
Table 1 Effect of temperature on rate and selectivity
| Temp./°C |
Time/h |
Isomer ratio 6
∶
7 |
| 4 |
168 |
95 ∶ 5 |
| 25 |
114 |
83 ∶ 17 |
| 60 |
10 |
67 ∶ 33 |
On reducing the reaction temperature from 60 to 4 °C the de improved from 43 to 90% but the reaction times became intolerably long, one week at 4 °C. However, performing the reaction at 4 °C gave an isolated yield of 73% for adduct 6 as a pure diastereoisomer. The diene 5 proved to be unstable to a range of p-block Lewis acid catalysts, boron trifluoride–diethyl ether, aluminium trichloride, and dimethylaluminium chloride, even at low temperatures, leaving limited scope for catalysis. Thermal conditions could not be found for effecting a Diels–Alder reaction of diene 5 with dienophiles activated with only one electron withdrawing group. Decomposition of diene 5 was observed on attempted reaction with ethyl acrylate in refluxing toluene.
5 M Lithium perchlorate in diethyl ether is known to catalyse Diels–Alder reactions.29,30 However, there was no data available on what effect this medium would have on regiochemistry or exo–endo selectivity. To gain insight into how N-dienes behave in this medium, the Diels–Alder chemistry of known achiral lactam diene 8 in ethereal 5 M lithium perchlorate solution was investigated. With dienes activated with two electron withdrawing groups, (E)-3-phenylsulfonylacrylic acid benzyl ester and dimethyl fumarate, Diels–Alder reaction did indeed take place with diene 8 after refluxing for several days. However, a 4 ∶ 1 and a 1 ∶ 1 mixture of isomers resulted respectively, and these dienophiles were not investigated further. However, when (E)-nitrostyrene was used as dienophile a Diels–Alder
reaction resulted which was completely regio- and endo-selective and gave adduct 9 in 64% yield as a single isomer. The regiochemistry was readily determined by coupling of 6′-H at δ 5.33, due to CHNO2, to 1′-H at δ 5.67, and the magnitude of the coupling constant J1′-H,6′-H of 6.0 Hz confirmed endo-stereochemistry. The large coupling constant J6′-H,5′-H 12.9 Hz confirmed that the stereochemistry of the alkene was preserved in the product and that both the nitro and phenyl groups were trans-diequatorial. 5 M Ethereal lithium perchlorate solution has previously been employed to catalyse Diels–Alder reactions using nitroalkenes as the dienophiles.31,32 Given the high selectivity of this reaction our attention turned to a similar study with chiral diene 5.
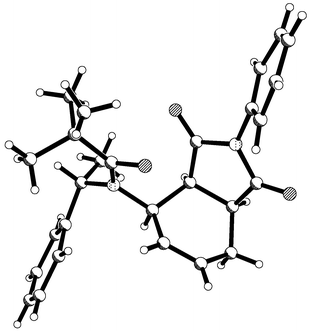
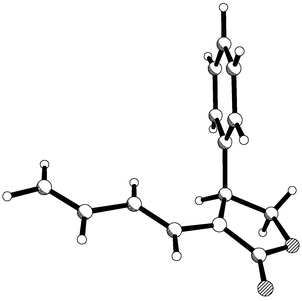
Chiral diene 5 reacted with (E)-nitrostyrene in 5 M lithium perchlorate solution at 35 °C for four days and gave two adducts 10 and 11 (ratio 17 ∶ 83) from which adduct 11 could be isolated in 61% yield by crystallisation. Single crystal X-ray crystallography was used to determine the absolute configuration of adduct 11 and this is depicted in Fig. 2. Again the stereochemistry of the major adduct was consistent with the carbonyl group taking up a conformation anti to the diene, as drawn, and the dienophile then approaching in an endo-fashion from the sterically less hindered face. Although this result was very encouraging, the de value for the cycloaddition was still below synthetically useful levels.
Clearly, to get better de values in this reaction it was necessary to have something larger than a methyl group on the auxiliary. Unfortunately, at the time of this study optically pure starting materials for varying the group on the 3-alkylisoindolin-1-one auxiliary were not readily available, and furthermore, the ortho-lithiation reaction employed in making the auxiliary was very inefficient. A new readily available auxiliary was sought. However, it should be noted that new efficient approaches to chiral 3-substituted isoindolin-1-ones have recently been published.33
 |
| | Fig. 2 X-Ray structure of major adduct 11 derived from the Diels–Alder reaction depicting absolute stereochemistry. | |
Preparation.
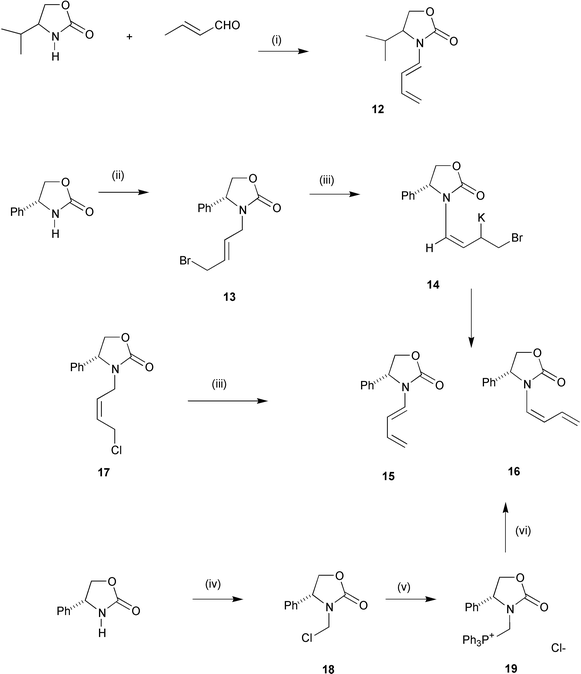
Oxazolidinones were next investigated as auxiliaries, as a wide range of the precursor amino alcohols were readily available in both enantiomerically pure forms. Racemic 4-isopropyloxazolidin-2-one readily condensed with crotonaldehyde and gave diene 12 in 52% yield. Unfortunately, the reaction of (R)-(−)-4-phenyloxazolidin-2-one with α,β-unsaturated aldehydes was capricious and although diene 15 was occasionally obtained, it was not reproducible and in most cases intractable material resulted. Although diene 15 was a known compound, it had only ever previously been isolated as a by-product.34 A new, more reliable approach to diene 15 had to be found. A dehydrobromination sequence was first investigated. Alkylation of
(R)-(−)-4-phenyloxazolidin-2-one with (E)-1,4-dibromobut-2-ene gave adduct 13 in 35% yield along with substantial amounts of a disubstituted product. Dehydrobromination of 13 with potassium tert-butoxide as base gave a 4 ∶ 1 mixture of Z- and E-dienes 16 and 15 respectively in 77% combined yield. The stereochemistry of these dienes was readily assigned by measurement of coupling constant J1′-H,2′-H. Chemical shifts and coupling constants for proton 1′-H for the Z- and E-dienes 16 and 15 were δ 6.04, J 8.8 Hz and δ 6.82, J 14.4 Hz respectively, unambiguously establishing
the stereochemistry of these two isomers. The E-diene 15 was clearly the thermodynamically more stable isomer indicating a kinetic preference for formation of the Z-isomer on elimination. One possible explanation was that the predominant mechanism for elimination was not E2′ as anticipated, but instead E1cB. Hence, deprotonation of 13 gave predominantly syn-π-allyl potassium intermediate 14, depicted (Scheme 3) as the σ-allyl isomer for clarity, stabilised by chelation to one of the heteroatoms in the oxazolidinone ring. The slow step was then elimination of bromide from this species which gave predominantly the Z-diene 16. When Z-alkene 17 was treated under the same elimination conditions,
again Z-diene 16 predominated, suggesting a common pathway and similar intermediate in both elimination sequences. Despite extensive experimentation, conditions could not be found to isomerise the Z-diene 16 to E-diene 15. The Z- and E-dienes could not be separated by flash chromatography. However, only the E-isomer 15 was reactive in the Diels–Alder reaction with N-phenylmaleimide and upon completion the pure Z-isomer could be readily separated from the cycloadduct. Conditions could not be found to effect a Diels–Alder reaction of Z-diene 16 with our best dienophile, N-phenylmaleimide. Presumably, with the very bulky N-auxiliary on the Z-diene, it is difficult
to adopt the prerequisite s-cis-conformation required for Diels–Alder reaction. Z-Dienes with oxygen substituents at the termini are known to participate in Diels–Alder reactions suggesting that the presence of a Z-heteroatom is not detrimental to Diels–Alder chemistry.35
 |
| | Scheme 3
Reagents and conditions: (i) PPTS, toluene, reflux; (ii) 1,4-dibromobutene, THF, KOH; (iii) KOBut, THF; (iv) trimethylsilyl chloride, THF, paraformaldehyde; (v) triphenylphosphine, acetonitrile; (vi) LDA, THF, then acrolein. | |
The second approach to these dienes involved Wittig chemistry. Phosphonium salt 19 was readily prepared in 73% yield by reaction of triphenylphosphine with (R)-(−)-N-chloromethyl-4-phenyloxazolidin-2-one. Wittig reaction of the ylide derived from this salt with acrolein gave a 4 ∶ 1 mixture of Z- and E-dienes 16 and 15 respectively in a poor 24% yield. No attempt was made to optimise this reaction as again the unwanted unreactive Z-diene was the major isomer produced.
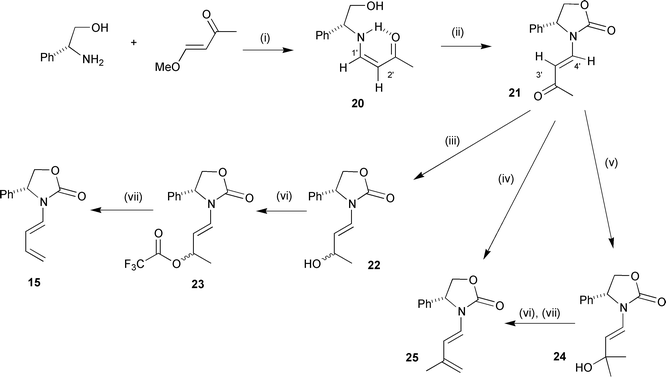
A successful reproducible approach to E-diene 15 involved assembling the E-enamide double bond first and then introducing the second double bond by an elimination reaction (Scheme 4). Hence, reaction of (R)-(−)-phenylglycinol with (E)-4-methoxybut-3-en-2-one gave exclusively the Z-enaminone 20 in 85% yield. The stereochemistry of the double bond was determined to be Z, based on the coupling constant J1′-H,2′-H of 7.4 Hz from the doublet for proton 2′-H, chemical shift δ 5.02. It is well documented that such enaminones form intramolecular hydrogen bonds with carbonyl groups, hence favouring the Z-alkene.36–38 Further evidence for this intramolecular hydrogen bond was the high chemical shift
for the NH proton at δ 10.31 and the extremely large vicinal coupling constant J1′-H,N–H of 12.6 Hz, which strongly implied that the dihedral angle between these two hydrogen atoms was large as illustrated in structure 20 (Scheme 4). Reaction of Z-enaminone 20 with boiling diethyl carbonate containing potassium carbonate gave the E-oxazolidinone 21 in 94% yield. The stereochemistry of adduct 21 was unambiguously assigned as E based on the coupling constant J3′-H,4′-H of 14.7 Hz. An analytical sample of 21 was obtained by recrystallisation from ethanol but analysis of the mother liquor showed a substantial quantity of enaminone 20 present. The vinylogous imide character of the oxazolidinone carbonyl group is clearly making this substrate susceptible to attack from the nucleophilic recrystallisation solvent, seriously reducing the yield. Crude material was used for the subsequent steps. Reduction of the ketone carbonyl group proceeded smoothly with sodium borohydride and gave a 1 ∶ 1 mixture of diastereoisomeric alcohols 22 in 99% crude yield. Initially, the E-diene 15 was prepared by conversion of the secondary alcohol to an acetate followed by palladium catalysed elimination of this group. However, this reaction was slow and never went to completion. In an attempt to optimise these final steps a trifluoroacetate was employed, as it is known that allylic trifluoroacetates readily eliminate to give dienes under palladium catalysis.39 Serendipitously,
attempted formation of trifluoroacetate 23, followed by a non-aqueous work up resulted in substantial amounts of the E-diene 15 being produced. An NMR scale reaction was performed using acetonitrile-d3 as solvent and this revealed that by using pyridine as base with a catalytic quantity of 4-dimethylaminopyridine, the trifluoroacetate 23 was formed quantitatively in less time that it took to run the spectrum after addition of trifluoroacetic anhydride (approximately five minutes). The trifluoroacetate 23 proved to be very sensitive and did not survive a mildly basic aqueous work up. Monitoring the NMR scale reaction revealed that the trifluoroacetate 23 was stable at 25 °C under the reaction conditions, but on heating to 80 °C new signals for the diene 15 appeared in the proton NMR spectrum. Addition of 2.3 molar equivalents of triethylamine and heating to 80 °C for two hours resulted in a clean elimination and gave the E-diene 15 as the sole reaction product, eliminating the need to handle the sensitive trifluoroacetate 23. There appears to be no literature precedent for this simple elimination reaction. However, it is known that trifluoroacetates derived from amidic cyclic N,O-hemiacetals readily eliminate trifluoroacetic acid when treated with triethylamine to give cyclic enamides.40 Compound 23 is a trifluoroacetate of a vinylogous N,O-hemiacetal and as such it is not surprising that it should readily eliminate trifluoroacetic acid.
 |
| | Scheme 4
Reagents and conditions: (i) dichloromethane, 25 °C, 3 h; (ii) diethyl carbonate, potassium carbonate, 140 °C; (iii) sodium borohydride, ethanol; (iv) methyltriphenylphosphonium iodide, potassium tert-butoxide, THF; (v) methylmagnesium iodide, Et2O; (vi) pyridine, trifluoroacetic anhydride, acetonitrile, 25 °C, 5 min; (vii) triethylamine, acetonitrile, 80 °C, 2 h. | |
Conditions from the NMR scale reaction were used to reproducibly synthesise the crystalline E-diene 15 on a preparative scale in 81% yield. This dehydration methodology could also be extended to tertiary allylic alcohol 24, produced in 74% yield by reaction of enaminone 21 with methylmagnesium iodide and gave 3′-methyldiene 25 in 59% overall yield. This procedure was much more efficient than a one pot Wittig reaction which gave diene 25 in an unsatisfactory 33% yield from ‘ketone’
21.
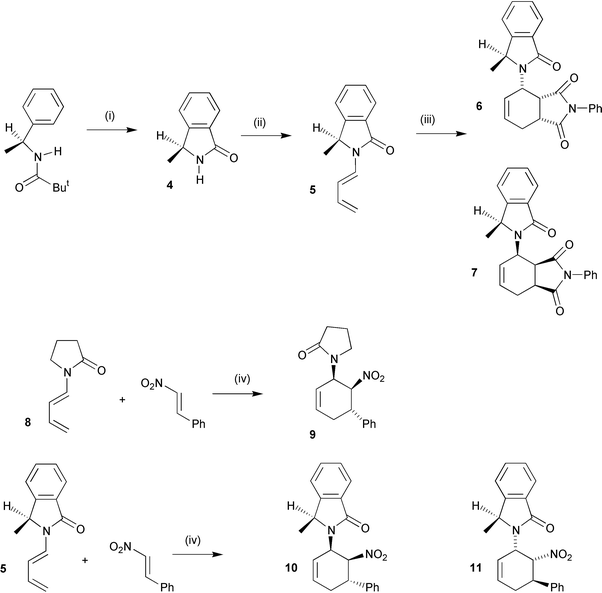
Fig. 3 shows the X-ray structure of crystalline diene 15. It clearly shows that in the solid state the diene adopts an s-trans-conformation with the carbonyl group of the auxiliary anti to the diene. The model developed for predicting the diastereoselectivity on cycloaddition in solution required that the carbonyl group of the auxiliary was anti to the diene, indicating that the predictive model may well have real physical significance. Table 2 tabulates the chemical shifts of the alkene protons for all of the dienes that were prepared. For the range of auxiliaries employed, there is very little variation in the chemical shift values for protons 3′-H and 4′-H (range 0.4 ppm), indicating that the electronic influence of the nitrogen is similar in all of these substrates. However, there is a relatively large spread in chemical shift value for
protons 1′-H and 2′-H, both in the range 0.7 ppm, indicating that additional through-space interactions from the auxiliary are coming into play. Of particular note is that with the unsubstituted auxiliary 5 (Table 2, entry 2), proton 2′-H has its highest chemical shift value of 5.9 ppm. On going to an oxazolidinone with a 4-isopropyl group this value decreases to 5.6 ppm. On replacing the 4-isopropyl group with a phenyl group the chemical shift of 2′-H decreases by 0.4 to its lowest value of 5.2 ppm. This trend indicates that there is a through-space interaction between proton 2′-H and the pendant aromatic ring, strongly suggesting that the anti-conformer depicted in the crystal structure is also the most favoured conformer in solution. The chemical shift values for proton 1′-H for the E-dienes are in the range that is expected from empirical calculations using
additive shielding factors. However, the same calculations suggest that proton 1′-H for the Z-isomer 16 should have a chemical shift of 0.15 ppm higher than that for the E-isomer 15! Measured chemical shift values clearly show that proton 1′-H for the E-diene 15 has a chemical shift value of 0.8 ppm higher than that for the Z-diene 16. The X-ray structure of diene 15 (Fig. 3) clearly shows that proton 1′-H is in the deshielding cone of the carbamate carbonyl group. The empirical calculation is probably allowing for the strong anisotropic interaction of the carbonyl group with proton 1′-H for both the E- and Z-dienes. Clearly with the Z-diene 16,
with the bulky auxiliary, a major conformational change has taken place removing proton 1′-H from the carbonyl shielding zone. This conformational change is probably a contributing factor to the lack of reactivity of diene 16.
Table 2 Variation in diene chemical shift as a function of auxiliary
| |
Chemical shift values for diene protons |
| Diene |
1′-H |
2′-H |
3′-H |
4′-H |
|
1
|
6.6 |
5.6 |
6.2 |
4.98/5.01 |
|
5
|
7.3 |
5.9 |
6.4 |
5.0/5.2 |
|
8
|
7.1 |
5.6 |
6.4 |
5.0/5.1 |
|
12
|
6.7 |
5.6 |
6.2 |
4.9/5.1 |
|
15
|
6.8 |
5.2 |
6.1 |
4.79/4.81 |
|
25
|
6.8 |
5.4 |
|
4.6/4.8 |
|
Z-16 |
6.0 |
5.5 |
6.3 |
5.0/5.1 |
 |
| | Fig. 3 X-Ray structure of diene 15 depicting the solid state conformation in which the carbamate carbonyl group is anti to the diene. | |
Diels–Alder reactions of 4-substituted oxazolidin-2-one dienes.
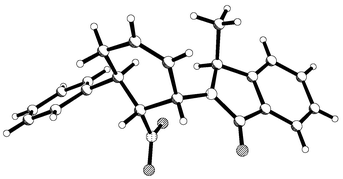
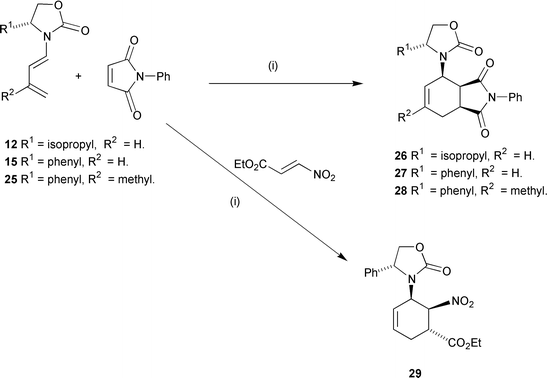
Reaction of dienes 12, 15 and 25 with N-phenylmaleimide gave exclusively endo-adducts 26, 27 and 28 in 67, 75, and 69% yield respectively after recrystallisation (Scheme 5). On examination of the crude 13C NMR spectra of these adducts additional small peaks at less than 2% intensity were observed, which were tentatively assigned to the other endo-diastereoisomer. In all cases the major isomer was ready purified by crystallisation and single crystal X-ray analysis on adduct 27 (Fig. 4)
revealed that the relative and absolute configurations of these adducts were as shown. Again, the now well established model that has been developed correctly predicted the stereochemistry of this adduct. Because of the increased size of the group on the auxiliary, selective shielding of one face of the diene is now much more effective and the de value for this diene is substantially improved over that for diene 5. As before, the oxazolidinone dienes proved to be very sensitive to p-block Lewis acids ruling out these reagents as catalysts.
 |
| | Scheme 5
Reagents and conditions: (i) chloroform, room temperature. | |
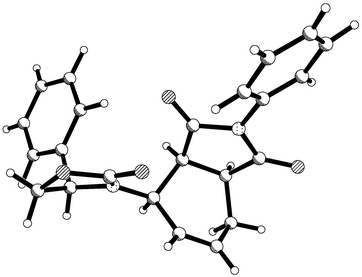 |
| | Fig. 4 X-Ray structure of endo-adduct 27 showing absolute stereochemistry. | |
Surprisingly dienes 15 and 25 proved to be unreactive towards nitrostyrene in boiling 5 M lithium perchlorate solution with the latter substrate decomposing under the reaction conditions. However, with the highly activated ethyl nitroacrylate, cycloaddition with diene 15 rapidly took place rapidly in chloroform at room temperature and gave two isomers (ratio 10 ∶ 1 by proton NMR spectroscopy), from which the pure adduct 29 was isolated in 65% yield after crystallisation.
4-Phenyloxazolidin-2-one auxiliaries are resistant to cleavage by hydrogenolysis and the most popular method for achieving this task employs sodium or lithium in liquid ammonia.41–44 Clearly, this procedure was incompatible with the highly oxidised functionality present in molecules 26–29. The Fischer method for oxazolidinone cleavage,45 using trimethylsilyl iodide looked ideal, but in our hands with substrate 27 this led only to intractable material. Unfortunately removal of the auxiliaries in these substrates is not a trivial task.
In conclusion, we have demonstrated that 4-phenyloxazolidin-2-ones and isoindolin-1-ones are effective groups for directing the stereochemical outcome of the Diels–Alder reaction of N-dienes. A model for predicting the stereochemical outcome of the cycloadditions has also been developed.
Experimental
General
Melting points were recorded using a Kofler hot stage apparatus and are uncorrected. IR spectra were recorded on a Perkin-Elmer Model 983G instrument coupled to a Perkin-Elmer 3700 Data Station as potassium bromide (KBr) disks, or films (liquids). Proton nuclear magnetic resonance (NMR) spectra were recorded at 300 and 500 MHz using General Electric QE 300, Bruker DPX 300 and DRX 500 NMR spectrometers. Chemical shifts are given in parts per million (δ) downfield from tetramethylsilane as internal standard and coupling constants are given in hertz. The following abbreviations are used: s singlet, d doublet, t triplet, q quartet, m multiplet and br broad. Mass spectra were recorded using a Double Focusing Triple Sector VG Auto Spec and accurate molecular masses were determined by the peak matching method using perfluorokerosene as the standard reference and were accurate to within ±0.006 ppm. Microanalyses were obtained using a Perkin Elmer 2400 CHN elemental analyser.
Optical rotations were determined on a Perkin-Elmer precision polarimeter Model 241, using the specified solvent and concentration at the D-line 589 nm and at ambient temperature; values are reported in units of 10−1 deg cm2 g−1. Analytical TLC was carried out on Merck Kieselgel 60254 plates and the spots visualised using a Hanovia Chromatolite UV lamp. Flash chromatography was effected using Merck Kieselgel 60 (230–400 mesh). The solvents for which the Rf values are quoted are the same as those that were used for any subsequent chromatography.
X-Ray crystallography
Experimental and crystal data are listed in Table 3. Data for 15 and 27 were collected on a Bruker SMART diffractometer using the SAINT-NT46 software with graphite monochromated Mo-Kα radiation using phi/omega scans. Data for 11 were collected on a Siemens P4 diffractometer using the XSCANS software47 with graphite monochromated Mo-Kα radiation using omega scans. A crystal was mounted on to the diffractometer at low temperature under dinitrogen at ca. 120 K and the crystal stability for 15 and 27 was monitored via recollection of the first set of frames. For 11 the crystal stability was monitor by collection
of 3 standard reflections in every 100. There were no significant variations (< ±1%). Cell parameters were obtained from 1959, 2996 and 40 accurately centred reflections in the θ range 2–28° for 15 and 27, and 2–12.5° for 11 respectively. Lorentz and polarisation corrections were applied.
Table 3 Data collection and structure refinement details for 11, 15 and 27
| Compound |
11
|
15
|
27
|
| Empirical formula |
C23H24N2O4 |
C13H13NO2 |
C23H20N2O4 |
|
M
|
392.44 |
215.24 |
388.41 |
| Crystal size/mm |
0.52 × 0.42 × 0.21 |
0.45 × 0.27 × 0.18 |
0.22 × 0.20 × 0.09 |
| Crystal system |
Orthorhombic |
Orthorhombic |
Monoclinic |
| Space group (Z) |
P212121 (4) |
P212121 (4) |
P21 (4) |
|
a/Å |
25.491(2) |
6.0693(6) |
9.6451(7) |
|
b/Å |
7.795(1) |
8.7995(9) |
9.5458(7) |
|
c/Å |
9.982(1) |
21.691(2) |
9.9442(8) |
|
β/° |
— |
— |
99.358(2) |
|
U/Å3 |
1983.4(4) |
1158.4(2) |
903.38(12) |
|
D
c/g cm−3 |
1.314 |
1.234 |
1.428 |
|
F(000) |
832 |
456 |
408 |
|
μ(Mo-Kα)/mm−1 |
0.090 |
0.084 |
0.099 |
| 2θ range/° |
4–50 |
3.5–57 |
4–58 |
| Unique reflections |
2355 |
2405 |
3890 |
|
wR2 (R1) |
0.0969 (0.0407) |
0.1306 (0.0483) |
0.1080 (0.0515) |
The structures were solved using direct methods and refined with the SHELXTL program package48 and the non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen-atom positions were located from difference Fourier maps and then fully refined for 15 and 27. Hydrogen atoms were added at idealised positions for 11 and a riding model was used for subsequent refinement. The ethyl acetate solvate is disordered over two symmetry related positions. The absolute configurations for 11, 15 and 27 were assigned based on the known configuration of the auxiliary. The function minimised was Σ[w(|Fo|2
−
|Fc|2)]
with reflection weights w−1
= [σ2|Fo|2
+ (g1P)2
+ (g2P)] where P
= [max |Fo|2
+ 2|Fc|2]/3. Additional material available from the Cambridge Crystallographic Data Centre comprises relevant tables of atomic coordinates, bond lengths and angles, and thermal displacement parameters.†
Preparation of dienes
(1′E,1″S)-(+)-2,2-Dimethyl-N-(buta-1′,3′-dienyl)-N-(1″-phenylethyl)propionamide 1.
(S)-α-Methylbenzylamine (4.7 g, 3.9 mmol) was added to a solution of crotonaldehyde (2.7 g, 3.9 mmol) in dichloromethane (20 ml) and the resulting mixture was stirred at room temperature for 1 h. The mixture was put in a separating funnel and the water layer removed. Magnesium sulfate (2 g) was added and the solution was stirred at room temperature for a further 1 h whereupon the magnesium sulfate was removed by filtration and the solution concentrated to give an oil. This oil was dissolved in benzene (45 ml) containing triethylamine (4.6 g, 4.6 mmol) and trimethylacetyl chloride (5.5 g, 4.6 mmol) was added dropwise over 5 min to this solution. The solution was refluxed for 2 h, cooled, washed with saturated sodium bicarbonate solution (2 × 20 ml),
water (20 ml), dried over sodium sulfate and concentrated to give a red oil. Kugelrohr distillation gave the title compound (8.2 g, 82%) as a pale yellow oil, bp 150 °C at 0.1 mmHg; [α]D
=
+93.2 (c 0.99, CHCl3) (C17H23NO requires C, 79.3; H, 9.0; N, 5.4%. Found C, 79.1; H, 8.9; N, 5.4%); νmax (KBr)/cm−1 3087, 3030, 2975, 1631, 1151, 1001, 698; δH (500 MHz, CDCl3) 1.34 (9H, s, 3 × CH3), 1.62 (3H, d, J 7.0, CHCH3), 4.98 (1H, d, J 10.3, 4′-H), 5.01 (1H, d, 17.4, 4′-H), 5.59 (1H, dd, J 14.2, 10.3, 2′H), 5.98 (1H, q, J 7.0, CHCH3), 6.21 (1H, dt, J 17.4, 10.3, 3′-H), 6.56 (1H, d, J 14.2, 1′H), 7.27 (3H, m, Ar-H), 7.34 (2H, m, Ar-H); δH
(125 MHz, CDCl3) 16.2, 27.6, 40.3, 53.3, 116.2, 122.9, 126.3, 126.6, 127.2, 128.3, 134.4, 141.2, 177.3; m/z (%) 257 (M+, 75), 205 (83), 154 (84), 138 (81), 104 (96), 51 (100).
(1S)-(+)-N-(2,2-Dimethylpropionyl)-1-phenylethylamine.
Trimethylacetyl chloride (5.47 g, 45 mmol) was added dropwise over 10 minutes to a magnetically stirred solution of (S)-(−)-methylbenzylamine (5.0 g, 41 mmol) and triethylamine (4.58 g, 45 mmol) in dichloromethane (30 ml) at 0 °C. When the addition was complete the reaction was allowed to warm to room temperature and stirred for a further 16 h. Water (10 ml) was added and the aqueous layer was extracted with dichloromethane (30 ml). The combined organic layers were washed with saturated sodium bicarbonate solution dried over magnesium sulfate and concentrated to give a white solid. Recrystallisation from hexane–dichloromethane gave the title compound (7.72 g, 92%) as white needles, mp 117–118 °C, lit. mp for enantiomer 115–117 °C;28 [α]D
=
+107.6 (c 0.8, EtOH)
(C13H19NO requires C, 76.1; H, 9.3; N, 6.8%. Found C, 75.9; H, 9.6; N, 6.9%); νmax (KBr)/cm−1 3340, 1638; δH (500 MHz, CDCl3) 1.20 (9H, s, 3 × CH3), 1.48 (3H, d, J 6.9, CH-CH3), 5.12 (1H, m, CH-CH3), 5.84 (1H, br s, NH), 7.24–7.36 (5H, m, 5 × Ar-H); δC (125 MHz, CDCl3) 13.1, 45.7, 115.1, 116.5, 120.9, 124.6, 124.7, 141.9, 164.3; m/z (%) 205 (M+, 36), 190 (6), 105 (100), 77 (19), 57 (64).
(3S)-(−)-3-Methylisoindolin-1-one 4.
n-Butyllithium (9.1 ml, 1.6 M in hexane) was added dropwise to a solution of (S)-(+)-N-(2,2-dimethylpropionyl)-1-phenylethylamine (3.0 g, 15 mmol) in dry THF (30 ml) under a nitrogen atmosphere at 0 °C and the resulting solution was cooled to −78 °C. tert-Butyllithium (10.3 ml, 1.7 M in pentane) was added to the resulting solution and this was allowed to warm to −60 °C, recooled to −78 °C and diethyl carbonate (2.58 g, 22 mmol) was added dropwise. The resulting stirred solution was allowed to warm to room temperature, water (20 ml) was cautiously added and the resulting mixture was stirred for a further 30 minutes. THF was removed under reduced pressure and the aqueous layer was extracted with dichloromethane (2 × 30 ml). The combined organic layers were dried over magnesium sulfate and concentrated
under reduced pressure. Flash chromatography gave the title compound (0.52 g, 24%) as a white solid, mp 102–103 °C, Rf 0.13 (ether); [α]D
=
−39.8 (c 0.6, EtOH) (C9H9NO requires C, 73.4; H, 6.2; N, 9.5%. Found C, 73.1; H, 6.5; N, 9.3%); νmax (KBr)/cm−1 3192, 1617; δH (500 MHz, CDCl3) 1.51 (3H, d, J 6.8, CH3), 4.71 (1H, q, J 6.8, CH-CH3), 7.44 (2 × 1H, 2 × m, 2 × Ar-H), 7.56 (1H, m, 1 × Ar-H), 7.85 (1H, d, J 7.6, Ar-H), 7.93 (1H, br s, NH); δC (125 MHz, CDCl3) 13.1, 45.7, 115.1, 116.5, 120.9, 124.6, 124.7, 141.9, 164.3; m/z (%) 147 (M+, 50), 132 (100), 104 (24), 77 (23).
(3S,1′E)-(−)-N-2-(Buta-1′,3′-dienyl)-3-methylisoindolin-1-one 5.
A solution of (3S)-(−)-3-methylisoindolin-1-one (0.27 g, 1.84 mmol), freshly distilled crotonaldehyde (0.38 ml, 4.61 mmol) and pyridinium toluene-p-sulfonate (0.1 g, 0.4 mmol) in dry benzene (40 ml) was boiled for 3 h with azeotropic removal of water using a Dean–Stark trap. Concentration of the resulting solution gave a yellow oil. Flash chromatography gave the title compound (0.23 g, 62%) as a clear oil, Rf 0.8 (ether); [α]D
=
−10.8 (c 0.5, EtOH) (C13H13NO requires 199.0997. Found M+ 199.0999); νmax (film)/cm−1 1700, 1646; δH (500 MHz, CDCl3) 1.57 (3H, d, J 6.6, CH3),
4.84 (1H, q, J 6.6, CHN), 5.04 (1H, d, J 10.4, 4′-H), 5.20 (1H, d, J 16.9, 4′-H), 5.88 (1H, dd, J 10.5, 14.4, 2′-H), 6.44 (1H, dt, J 10.4, 16.9, 3′-H), 7.27 (1H, d, J 14.7, 1′-H), 7.44–7.58 (2 × 1H, 2 × m, 4-H, 6-H), 7.62 (1H, dd, J 7.4, 0.9, 8-H), 7.88 (1H, d, J 7.4, 7-H); δC (125 MHz, CDCl3) 19.1, 54.9, 113.7, 114.5, 122.1, 124.2, 125.5, 128.5, 132.6, 135.3, 147.2; m/z (%) 199 (M+, 100), 184 (66), 170 (24), 104 (30), 77 (35).
(1E)-N-Buta-1,3-dienylpyrrolidin-2′-one 8.
A solution of pyrrolidin-2-one (20.0 g, 0.24 mol), freshly distilled trans-crotonaldehyde (16.5 g, 0.24 mol) and pyridinium toluene-p-sulfonate (0.3 g, 1.2 mmol) in dry toluene (200 ml) was boiled for 1 h with azeotropic removal of water using a Dean–Stark trap. The solution was cooled to ambient temperature, washed successively with saturated sodium bicarbonate solution (50 ml), water (50 ml), dried over magnesium sulfate and concentrated to give a heavy oil. After trituration with ether, the ether extract was concentrated to give the title compound (10.5 g, 33%) as a pale yellow solid, mp 58–59 °C, lit. mp 59.5–61.5 °C;12δH (300 MHz, CDCl3) 2.15 (2H, m, 4-H), 2.51 (2H, t, J 8.2, 3-H), 3.57 (2H, t, J
7.2, 5-H), 4.99 (1H, d, J 10.3, 4′-H), 5.14 (1H, d, J 16.2, 4′-H), 5.63 (1H, dd, J 14.3, 10.6, 2′-H), 6.35 (1H, m, 3′-H), 7.11 (1H, d, J 14.3 Hz, 1′-H).
(1′E)-3-Buta-1′,3′-dienyl-4-isopropyloxazolidin-2-one 12.
A solution of racemic 4-isopropyloxazolidin-2-one (2.0 g, 16 mmol), freshly distilled trans-crotonaldehyde (2.2 g, 31 mmol) and pyridinium toluene-p-sulfonate (0.1 g, 0.3 mmol) in toluene (100 ml) was heated for 5 h with azeotropic removal of water using a Dean–Stark trap. The solution was concentrated and flash chromatography gave the title compound (1.5 g, 52%) as a clear oil, Rf 0.57 (ether) (C10H15NO2 requires M+ 181.1103. Found M+ 181.1107); νmax (film)/cm−1 1750; δH (500 MHz, CDCl3) 0.84 (3H, d, J 6.8, CH3), 0.92 (3H, d, J 7.3, CH3), 2.19 (1H, m, (CH3)2CH),
4.19 (2 × 1H, 2 × m, 4-H and 5-H), 4.32 (1H, m, 5-H), 4.97 (1H, d, J 10.3, 4′-H), 5.10 (1H, d, J 17.6, 4′-H), 5.64 (1H, dd, J 14.7, 10.8, 2′-H), 6.32 (1H, m, 3′-H), 6.78 (1H, d, J 14.7, 1′-H); m/z (%) 181 (M+, 49), 138 (28), 130 (100), 121 (37), 86 (64).
(2E,4R)-(−)-N-(4-Bromobut-2-enyl)-4-phenyloxazolidin-2-one 13.
A suspension of finely ground potassium hydroxide (0.34 g, 6.13 mmol), (4R)-(−)-4-phenyloxazolidin-2-one (1.0 g, 6.13 mmol), triethylbenzylammonium chloride (0.07 g, 0.31 mmol) and (E)-1,4-dibromobutene (2.62 g, 12 mmol) in dry benzene (50 ml) was boiled under reflux for 12 h. The solution was allowed to cool to room temperature, washed with water (2 × 10 ml), dried over magnesium sulfate and concentrated to yield an orange oil. Flash chromatography gave the title compound (0.63 g, 35%) as a clear oil, Rf 0.54 [90 ∶ 10 ether–petroleum ether (40–60)]; [α]D
=
−50.7 (c 1.5, CHCl3) (C13H14NO2Br requires M+ 296.0286. Found M+ 296.0281); νmax
(film)/cm−1 2969, 2917, 1752, 735; δH (300 MHz, CDCl3) 3.26 (1H, dd, J 15.4, 6.8, 1 of N-CH2), 3.88 (2H, d, J 5.3, CH2-Br), 4.10–4.16 (2H, 2 × m, 1 of N-CH2, and CHHO), 4.63 (1H, t, J 8.8, PhCHN), 4.79 (1H, dd, J 7.1, 8.8 Hz, CHH-O), 5.64–5.68 (2 × 1H, 2 × m, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.29–7.44 (5H, m, 5 × Ar-H); m/z (%) 296 (M+, 0.3), 216 (100), 104 (35), 91 (80).
CH), 7.29–7.44 (5H, m, 5 × Ar-H); m/z (%) 296 (M+, 0.3), 216 (100), 104 (35), 91 (80).
Collection of further fractions yielded a white solid which was recrystallised from ethyl acetate–hexane and gave 1,4-bis[(2E,4R)-4-phenyl-2-oxooxazolidin-3-yl]but-2-ene (0.59 g, 25%) as white prisms, mp 143–144 °C; [α]D
=
−77.3 (c 0.4, CHCl3) (C22H22N2O4 requires C, 69.8; H, 5.9, N, 7.4%. Found C, 69.7; H, 5.7; N, 7.3%); νmax (film)/cm−1 2927, 2915, 2897, 1740, 1742; δH (300 MHz, CDCl3) 3.26 (2H, ddd, J 14.9, 4.2, 2.0, 2 × NCHH), 3.99 (2H, m, 2 × NCHH), 4.14 (2H, dd, J 8.3, 6.6, 5-H), 4.64 (2H, t, J 8.6, 4-H), 4.73 (2H, dd, J 8.8, 6.6, 5-H), 5.30 (2H, m, HCCH), 7.24–7.42 (10H, m, Ar-H); m/z (%)
334 (7.2), 215 (98), 158 (96), 104 (100), 91 (99).
(4R,1′Z)-(−)-3-(Buta-1′,3′-dienyl)-4-phenyloxazolidin-2-one 16.
Potassium tert-butoxide (92 mg, 0.82 mmol) was added to a solution of (4R,2′E)-(−)-3-(4′-bromobut-2′-enyl)-4-phenyloxazolidin-2-one (100 mg, 0.41 mmol) in dry THF (10 ml) and the resulting solution was stirred for 1 h at room temperature. THF was removed under reduced pressure, water (10 ml) was added and the residue extracted with dichloromethane (2 × 10 ml), dried over magnesium sulfate and concentrated to give a brown oil. Flash chromatography gave the title compound (68 mg, 77%) as a mixture of Z and E isomers. The Z-isomer was obtained pure by dissolving the mixture in chloroform (5 ml), adding N-phenylmaleimide (24 mg, 0.14 mmol) and stirring at room temperature overnight. Flash chromatography gave the title compound
(45 mg, 51%) as a clear oil, Rf 0.69 (ether); [α]D
=
−43.2 (c 0.5, CHCl3) (C13H13NO2 requires M+ 215.0946. Found M+ 215.0943); νmax (film)/cm−1 2961, 2927, 1763; δH (500 MHz, CDCl3) 4.19 (1H, dd, J 8.6, 4.8, 4-H), 4.71 (1H, t, J 8.6, 5-H), 5.02 (1H, d, J 9.9, 4′-H), 5.12 (1H, d, J 16.7, 4′-H), 5.23 (1H, dd, J 4.8, 8.6, 5-H), 5.52 (1H, dd, J 9.5, 11.4, 2′-H), 6.04 (1H, d, J 8.8, 1′-H), 6.40 (1H, m, 3′-H), 7.23–7.27 (2H, m, 2 × Ar–H), 7.34–7.42 (3H, m, 3 × Ar-H); m/z (%) 215 (M+, 100), 104 (90), 77 (36), 53 (84).
(4R)-(−)-3-Chloromethyl-4-phenyloxazolidin-2-one 18.
A solution of (4R)-(−)-4-phenyloxazolidin-2-one (0.5 g, 3.1 mmol) and paraformaldehyde (0.1 g, 3.4 mmol) in trimethylsilyl chloride (5 ml) was refluxed for 2 hours. The volatile materials were removed in vacuo and the remaining solid was recrystallised from ether–hexane to give the title compound (0.5 g, 77%) as a cream coloured solid, mp 48–50 °C; [α]D
=
−50.1 (c 0.9, CHCl3) (C10H10NO2Cl requires C, 56.8; H, 4.8; N, 6.6%. Found C, 56.6; H, 4.8; N, 6.3%); νmax (KBr)/cm−1 3015, 2977, 2917, 1768, 707; δH (500 MHz, CDCl3) 4.19 (1H, t, J 9.2, 4-H), 4.53 (1H, d, J 10.7, 1′-H), 4.71 (1H,
t, J 9.2, 5-H), 5.04 (1H, t, J 9.2, 5-H), 5.59 (1H, d, J 10.7, 1′-H), 7.36–7.38 (2H, m, Ar-H), 7.43–7.48 (3H, m, Ar-H); δC (125 MHz, CDCl3) 53.8, 57.5, 69.7, 126.9, 128.9, 129.2, 134.9, 156.5; m/z (%) 211 (M+, 2.3), 176 (100), 132 (35), 91 (76).
(4R)-(−)-(2-Oxo-4-phenyloxazolidin-3-ylmethyl)triphenylphosphonium chloride 19.
A solution of triphenylphosphine (3.5 g, 13.0 mmol) and (4R)-(−)-3-chloromethyl-4-phenyloxazolidin-2-one (1.4 g, 6.6 mmol) in acetonitrile (30 ml) was stirred overnight at room temperature. The solvent was removed under reduced pressure to yield a sticky solid which was dissolved in dichloromethane (4 ml) and then precipitated with ether to yield the title compound (2.3 g, 73%) as a white solid, mp 175–176 °C; [α]D
=
−72.8 (c 0.6, EtOH) (C28H25NO2PCl requires C, 71.0; H, 5.3; N, 3.0%. Found C, 70.8; H, 5.4; N, 2.9%); νmax (KBr)/cm−1 1768, 1440, 746, 689; δH (300 MHz, CDCl3) 4.15 (1H, dd, J 8.6, 6.0, 4-H),
4.74 (1H, t, J 8.6, 5-H), 5.24 (1H, dd, J 16.2, 1.5, 1′-H), 5.52 (1H, t, J 8.6, 5-H), 5.98 (1H, dd, J 16.2, 4.4, 1′-H), 7.61–7.82 (15H, m, 3 × Ar-H); m/z (%) 438 (M+
− 1, 14), 277 (100).
(4R)-(−)-N-(Buta-1′,3′-dienyl)-4-phenyloxazolidin-2-one.
Lithium diisopropylamide solution (0.9 ml, 1.5 M in hexane, 1.4 mmol) was added dropwise to a solution of (4R)-(−)-(2-oxo-4-phenyloxazolidin-3-ylmethyl)triphenylphosphonium chloride (0.5 g, 1.1 mmol) in dry THF (20 ml) at −78 °C under a nitrogen atmosphere. The resulting mixture was stirred for 15 minutes at this temperature and acrolein (74 mg, 1.1 mmol) was added dropwise and the resulting mixture was allowed to warm to room temperature. Water (20 ml) was added and the product extracted with dichloromethane (2 × 20 ml), dried over magnesium sulfate and concentrated. Flash chromatography gave a 4 ∶ 1 mixture of Z and E dienes 16 and 15 (58 mg, 24.0%) as a yellow oil. Data for diene 16
are already listed above and those for diene 15 are provided below.
(1′R,1Z)-(−)-4-(2′-Hydroxy-1′-phenylethylamino)but-3-en-2-one 20.
A solution of (R)-(−)-phenylglycinol (27.4 g, 0.2 mol) and 4-methoxybut-3-en-2-one (20.0 g, 0.2 mol) in dichloromethane (150 ml) was stirred at room temperature for 2 h. Removal of the solvent gave a viscous yellow oil which gave the title compound (34.8 g, 85%) as a white solid on trituration with hexane–ethanol (10 ∶ 1). This sample was deemed pure enough for the next stage. However, an analytical sample (mp 130 °C) was obtained by recrystallisation from aqueous methanol, [α]D
=
−24 (c 0.5, CHCl3) (C12H15NO2 requires C, 70.2; H, 7.4; N, 6.8%. Found C, 70.4; H, 7.2; N, 6.7%); νmax (KBr)/cm−1 2964, 1753, 1650, 1428, 1400, 1263, 1089,
1015, 802; δH (300 MHz, CDCl3) 1.98 (3H, s, 1-H), 2.92 (1H, br s, OH), 3.78 (1H, dd, J 11.5, 8.1, 2′-H), 3.87 (1H, dd, J 11.5, 4.4, 2′-H), 4.39 (1H, td, J 8.1, 4.4, 1′-H), 5.02 (1H, d, J 7.4, 3-H), 6.73 (1H, dd, J 12.6, 7.4, 4-H), 7.21–7.39 (5H, m, Ar-H), 10.32 (1H, br dd, J 12.6, 8.1, NH); δC (75 MHz, CDCl3) 28.5, 64.2, 66.1, 94.3, 126.3, 127.6, 137.5, 151.9, 190.2, 197.6; m/z (%) 205 (M+, 10), 174 (100).
(4R,1′E)-(−)-3-(3′-Oxobut-1′-enyl)-4-phenyloxazolidin-2-one 21.
Potassium carbonate (9.4 g, 68 mmol) was added to a solution of (1′R,1Z)-4-(2′-hydroxy-1′-phenylethylamino)but-3-en-2-one (26.0 g, 127 mmol) in diethyl carbonate (80 ml) and the apparatus was set up for distillation. The suspension was heated at 140 °C for 6 h whereupon ethanol collected in the receiving flask. The mixture was allowed to cool to room temperature, ethyl acetate (100 ml) was added and the resulting solution was washed with water (2 × 50 ml) and dried over magnesium sulfate. Addition of petroleum ether (40–60) precipitated the title compound (27.5 g, 94%) as an orange solid, mp 158–160 °C, lit. mp 161–162 °C.16 An analytical sample was obtained by recrystallisation from ethanol but as this led
to major losses the crude material was used for subsequent stages. [α]D
=
−185 (c 1.5, CHCl3) (C13H13NO3 requires C, 67.5; H, 5.7; N, 6.1%. Found C, 67.5; H, 5.9; N, 5.9%); νmax (KBr)/cm−1 3220, 3040, 2900, 1770, 1660, 1625, 1400, 1200, 760, 700; δH (300 MHz, CDCl3) 2.16 (3H, s, CH3), 4.23 (1H, dd, J 9.0, 4.8, 5-H), 4.80 (1H, t, J 9.0, 5-H), 5.06 (1H, dd, J 9.0, 4.8, 5-H), 5.27 (1H, d, J 14.7, 3′-H), 7.27 (2H, m, Ar-H), 7.41 (3H, m, Ar-H), 7.85 (1H, d, J 14.7, 4′-H); δC (75 MHz, CDCl3) 26.5, 58.0, 70.9, 112.3, 125.6, 128.7, 128.8, 136.4, 137.0, 154.9, 196.7; m/z (%) 231 (M+, 35), 174 (100).
(4R,3′R*)-3-(3′-Hydroxybut-1′-enyl)-4-phenyloxazolidin-2-one 22.
Sodium borohydride (1.2 g, 31.7 mmol) was added portionwise over 10 min to an ice-cooled solution of (4R,1′E)-(−)-3-(3′-oxobut-1′-enyl)-4-phenyloxazolidin-2-one (4.0 g, 17.3 mmol) in methanol (100 ml) and the resulting solution was stirred at room temperature for three hours. Water (15 ml) was added dropwise and the resulting solution was stirred for 15 minutes whereupon the solution was concentrated under reduced pressure. The residue was extracted with dichloromethane (2 × 20 ml), the combined extracts were dried over sodium sulfate and concentrated to give the title compound as a 1 ∶ 1 mixture of diastereoisomers (4.0 g, 99%) as a pale yellow oil. The two methyl doublets were the only peaks well resolved for the diastereoisomer mixture and these signals were
used to quantify the ratio of diastereoisomers. Due to the instability of these compounds the mixture was used directly without purification for the next step. δH (300 MHz, CDCl3) 1.10 (3H, d, J 6.2, 4′-H), 1.18 (3H, d, J 6.3, 4′-H), 4.20 (2H, m, 5-H), 4.25 (2H, m, 3′-H), 4.74 (4H, m, 5H and 2′H), 4.97 (2H, m, 5H), 6.81 (2H, d, J 14.5, 1′-H), 7.27 (4H, m, Ar-H), 7.38 (6H, m, Ar-H).
(4R,1′E)-(−)-3-(Buta-1′,3′-dienyl)-4-phenyloxazolidin-2-one 15.
Trifluoroacetic anhydride (3.2 g, 15.0 mmol) was added dropwise over 4 min to a stirred solution of (4R,3S*)-3-(3′-hydroxybut-1′-enyl)-4-phenyloxazolidin-2-one (2.0 g, 8.6 mmol), 4-dimethylaminopyridine (5 mg, 0.04 mmol) and pyridine (1.5 g, 19.0 mmol) in dry acetonitrile under a nitrogen atmosphere and the resulting solution was stirred at room temperature for 30 minutes. Triethylamine (2 g, 20.0 mmol) was added and the resulting mixture was boiled under reflux for 2 hours, allowed to cool to room temperature and the solvent removed under reduced pressure. Saturated sodium bicarbonate solution (20 ml) was added and the residue extracted with dichloromethane (2 × 20 ml). The combined dichloromethane extracts were dried over sodium sulfate, concentrated and purified by rapidly passing
through a short silica column using dichloromethane as eluant. Crystallisation from hexane gave the title compound as white needles (1.5 g, 81%), mp 100–101 °C, lit. mp 100–101 °C;34Rf 0.3 (ether); [α]D
=
−246 (c 5.3, CHCl3). The [α]D value in the original communication was erroneously reported as −43.3 (c 5.0 in EtOH)26 (C13H13NO2 requires C, 72.5; H, 6.1; N, 6.5%. Found C, 72.3; H, 6.0; N, 6.4%); νmax (KBr)/cm−1 2964, 1753, 1650, 1428, 1400, 1263, 1089, 1015, 802; δH (500 MHz, CDCl3) 4.07 (1H, dd, J 8.8, 5.2, 5-H), 4.66 (1H, t, J 8.8, 4-H), 4.79 (1H, d, J 10.4, 4′-H), 4.81 (1H, d, J 16.9, 4′-H),
4.97 (1H, dd, J 8.8, 5.2, 5-H), 5.24 (1H, dd, J 14.4, 10.6, 2′-H), 6.13 (1H, dt, J 16.9, 10.6, 3′-H), 6.82 (1H, d, J 14.4, 1′-H), 7.3–7.4 (5H, m, Ar-H); δC (75 MHz, CDCl3) 60.8, 72.9, 116.2, 116.9, 128.0, 128.2, 131.2, 131.7, 136.5, 140.1, 157.8; m/z (%) 215 (M+, 92), 170 (61), 156 (55), 104 (96), 91 (89), 80 (99), 53 (100).
(4R,1′E)-(−)-3-(3′-Hydroxy-3′methylbut-1′-enyl)-4-phenyloxazolidin-2-one 24.
Methyl iodide (0.5 g, 3.5 mmol) was added to a suspension of magnesium (0.1 g, 4.1 mmol) in ether (10 ml) and the resulting mixture was stirred at room temperature for 1 h. (4R,1′E)-(−)-3-(3′-Oxobut-1′-enyl)-4-phenyloxazolidin-2-one (0.5 g, 2.2 mmol) in THF (10 ml) was added in one portion to the Grignard solution and the resulting mixture was stirred at room temperature for 1 h. Water (20 ml) was cautiously added and the resulting solution was extracted with dichloromethane (3 × 10 ml), dried over magnesium sulfate and concentrated to give the title compound (0.4 g, 74%) as a clear oil. Attempted purification by flash chromatography led to decomposition so the material was used crude for the next step. δH (500 MHz, CDCl3)
1.31 (2 × 3H, 2 × s, 2 × CH3), 4.13 (1H, dd, J 8.7, 5.3, 5-H), 4.71 (1H, t, J 8.7, 4-H), 4.82 (1H, d, J 14.7, 2′-H), 4.99 (1H, dd, J 8.7, 5.3, 5-H), 6.81 (1H, d, J 14.7, 1′-H), 7.23–7.39 (5H, m, Ar-H).
(4R,1′E)-(−)-3-(3′-Methylbuta-1′,3′-dienyl)-4-phenyloxazolidin-2-one 25.
Potassium tert-butoxide (97 mg, 0.87 mmol) was added to a suspension of methyltriphenylphosphonium iodide (0.5 g, 1.3 mmol) in dry THF (20 ml). After stirring the mixture for 10 minutes, (4R,1′E)-(−)-3-(3′-oxobut-1′-enyl)-4-phenyloxazolidin-2-one (100 mg, 0.43 mmol) in tetrahydrofuran (2 ml) was added and the reaction mixture was stirred for 10 h at room temperature. Water (2 ml) was added and the volatiles were removed under reduced pressure. The aqueous layer was extracted with ether (2 × 10 ml), washed with brine, dried over magnesium sulfate, filtered and concentrated. Flash chromatography gave the title compound (32 mg, 32%), as a yellow solid, mp 69–70 °C, Rf 0.78 (ether); [α]D
=
−166.5
(c 0.6 EtOH) (C14H15NO2 requires M+ 229.1103. Found M+ 229.1113); νmax (KBr)/cm−1 3034, 2925, 1744, 1648; δH (500 MHz, CDCl3) 1.80 (3H, s, CH3), 4.15 (1H, dd, J 8.7, 5.0, 4-H), 4.63 (1H, d, J 1.1, 4′-H), 4.70–4.76 (2 × 1H, 2 × m, 4′-H, 5-H), 5.06 (1H, dd, J 9.1, 5.0, 5-H), 5.41 (1H, d, J 14.8, 2′-H), 6.82 (1H, d, J 14.8, 1′-H), 7.22–7.44 (5H, m, Ar-H); δC (125 MHz, CDCl3) 18.5, 58.4, 70.7, 114.6, 116.2, 122.9, 125.7, 128.4, 128.5, 128.7, 128.8, 129.4, 129.5, 133.7; m/z (%) 229 (M+, 96), 184 (26), 170 (41), 104 (100), 77 (28).
(3a′R,4′S,7a′R,1″S)-(−)-N-(1′,3′-Dioxo-2′-phenyl-2′,3′,3a′,4′,7′,7a′-hexahydro-1′H-isoindol-4′-yl)-2,2-dimethyl-N-(1″-phenylethyl)propionamide 3.
A solution of (1S,1′E)-N-(buta-1′,3′-dienyl)-(2″,2″-dimethylpropionyl)-1-phenylethylamine (0.50 g, 1.95 mmol) and N-phenylmaleimide (0.34 g, 1.95 mmol) in chloroform (50 ml) was refluxed for 24 hours. After cooling, the solvent was removed and the resulting oil was purified by flash chromatography to give the title compound (0.54 g, 64%) as a pale yellow solid, mp 78–79 °C, Rf 0.58 [50 ∶ 50, ether–petroleum ether (40–60)]; [α]D
=
−60 (c 0.7, CHCl3)
(C27H30N2O3 requires 430.2256. Found M+ 430.2258); νmax (film)/cm−1 2973, 2930, 1708, 1627; δH (500 MHz, CDCl3) 1.42 (9H, s, (CH3)3), 1.70 (3H, d, J 7.0, CH3), 1.91 (1H, m, 7′-H), 2.54 (1H, t, J 9.7, 3a′-H), 2.84 (1H, m, 7′-H), 2.97 (1H, m, 7a′-H), 3.48 (1H, d, J 9.5, 4′-H), 5.65 (1H, q, 6.9, CHCH3), 5.79 (2 × 1H, 2 × m, 5′-H, 6′-H), 7.27–7.49 (8H, m, Ar-H), 7.86 (2H, d, J 8.1, Ar-H); m/z (%) 430 (M+, 24), 345 (16), 241 (49), 105 (100), 79 (26), 57 (30).
(3a′S,4′R,7a′S,1″S)-(+)-N-(1′,3′-Dioxo-2′-phenyl-2′,3′,3a′,4′,7′,7a′-hexahydro-1′H-isoindol-4′-yl)-2,2-dimethyl-N-(1″-phenylethyl)propionamide 2.
Collection of further fractions gave a mixture of major and minor isomers from which the minor isomer 2 crystallised. Recrystallisation from ethanol gave the minor isomer 2 (0.11 g 13%) as prisms, mp 229–231 °C, Rf 0.52 [50 ∶ 50, ether–petroleum ether (40 ∶ 60)]; [α]D
=
+22 (c 1.0, CHCl3) (C27H30N2O3 requires C, 75.3; H, 7.0; N, 6.5%. Found C, 75.1; H, 6.9; N, 6.5%); νmax
(KBr)/cm−1 2980, 1706, 1629, 1178; δH (500 MHz, CDCl3) 1.35 (9H, s, (CH3)3), 1.94 (1H, m, 7′-H), 1.96 (3H, d, J 6.8, CHCH3), 2.85 (1H, ddd, J 16.1, 7.0, 2.2, 7′-H), 3.16 (1H, ddd, J 9.8, 6.0, 2.3, 7a′-H), 3.26 (1H, t, J 9.8, 3a′-H), 3.56 (1H, dq, J 10.1, 2.3, 4′-H), 5.62 (1H, q, J 6.8, CHCH3), 5.67 (1H, m, 6′-H), 5.76 (1H, dt, J 10.1, 2.3, 5′-H), 7.5–7.48 (10H, m, Ar-H). δH (125 MHz, CDCl3) 18.6, 20.8, 39.7, 40.5, 44.2, 51.6, 55.4, 122.4, 2 × 126.8, 2 × 126.9, 127.6, 128.2, 2 × 128.7, 2 × 128.8, 132,4, 132.8, 140.5, 175.9, 177.9, 178.0; m/z (%) 430 (M+ 13), 346 (12), 241 (50),
105 (100), 79 (41).
(3a′R,4′S,7a′R,3S)-(−)-2-(2′-Phenyl-1′,3′-dioxo-2′,3′,3a′,4′,7′,7a′-hexahydro-1′H-isoindol-4′-yl)-3-methylisoindolin-2-one 6.
A solution of N-phenylmaleimide (0.13 g, 0.75 mmol) and (3S,1′E)-(+)-N-2-(buta-1′,3′-dienyl)-3-methylisoindolin-1-one (0.15 g, 0.75 mmol) in chloroform (10 ml) was stored in a refrigerator at approximately 4 °C for 1 week. Concentration gave a heavy oil. Flash chromatography gave the title compound (0.21 g, 75%) as a heavy oil, Rf 0.15 (ether); [α]D
=
−14.33 (c 0.6, EtOH) (C23H20N2O3 requires M+ 372.1474. Found M+ 372.1479); νmax (film)/cm−1
3020, 1716, 755, 699; δH (500 MHz, CDCl3) 1.62 (3H, d, J 6.8, CHCH3), 2.38 and 3.03 (2 × 1H, 2 × m, 4′-H), 3.33 (1H, m, 7a′-H), 3.68 (1H, t, J 8.9, 3a′-H), 4.58 (1H, d, J 8.4, 7′-H), 4.7 (1H, q, J 6.5, CHCH3), 6.12 (2 × 1H, m, 5′-H and 6′-H), 7.25–7.51 (8H, m, 8 × Ar-H), 7.40–7.45 (2 × 1H, 2 × m, 4-H and 6-H), 7.54 (1H, m, 5-H), 7.79 (1H, d, J 7.6, 7-H); δC (125 MHz, CDCl3) 19.5, 22.5, 39.0, 42.6, 48.3, 58.1, 121.8, 123.4, 126.0, 126.5, 126.6, 128.0, 128.3, 128.7, 128.8, 128.9, 131.4, 131.6, 132.1, 147.3, 168.7, 175.7, 178.4; m/z (%) 372 (M+, 28), 199 (49), 146 (100), 80 (87).
(1′R*,5′S*,6′S*)-1-(6′-Nitro-5′-phenylcyclohex-2′-enyl)pyrrolidin-2-one 9.
A solution of (1′E)-N-(buta-1′,3′-dienyl)pyrrolidin-2-one (0.50 g, 3.7 mmol) and (E)-β-nitrostyrene (0.60 g, 4.0 mol) in 5 M ethereal lithium perchlorate solution (4 ml) was stirred for 1 week at room temperature. Water (10 ml) was added and the solution was extracted with dichloromethane (2 × 10 ml), dried over magnesium sulfate and concentrated to give a solid. Recrystallisation from ethanol gave the title compound (0.67 g, 64%) as a pale yellow solid, mp 178–179 °C (C16H18N2O3 requires M+ 286.1317. Found M+ 286.1328); νmax (KBr)/cm−1 1692, 1550, 1373; δH (500 MHz, CDCl3)
2.08 (1H, m, 3′-H), 2.15 (1H, m, 3′H), 2.40–2.55 (3 × 1H, 3 × m, 4-H, 2′H), 2.73 (1H, dt, J 18.4, 5.6, 4-H), 3.59–3.67 (2 × 1H, 2 × m, 5-H and 4′-H), 3.83 (1H, m, 4′-H), 5.33 (1H, dd, J 12.9, 6.0, 6-H), 5.65 (1H, m, 1-H), 5.72 (1H, m, 2-H), 6.25 (1H, m, 3-H), 7.33–7.44 (5H, m, Ar-H); m/z (%) 286 (M+, 0.15), 256 (0.75), 240 (64), 155 (71), 137 (100), 86 (91).
(3S,1′S,5′R,6′R)-(+)-3-Methyl-2-(6-nitro-5-phenylcyclohex-2-enyl)isoindolin-1-one 11.
A solution of (3S,1′E)-(+)-N-(buta-1,3-dienyl)-3-methylisoindolin-1-one (50 mg, 0.25 mmol) and (E)-β-nitrostyrene (37 mg, 0.25 mmol) in 5 M ethereal lithium perchlorate (4 ml) was stirred at 40 °C for 5 days. Water (10 ml) was added and the solution was extracted with dichloromethane (2 × 10 ml), dried over magnesium sulfate and concentrated to give a brown oil. Flash chromatography followed by crystallisation from ethyl acetate–hexane gave the title compound (53 mg, 61%) as white prisms, mp 64–65 °C, Rf 0.59 (ether); [α]D
=
+13.0 (c 0.4, EtOH) (C21H20N2O3 requires C, 72.4; H, 5.7, N,
8.0%. Found C, 72.1; H, 5.9; N, 8.3%); νmax (KBr)/cm−1 3055, 2981, 2932, 1692, 1619; δH (500 MHz, CDCl3) 1.57 (3H, d, J 6.7, CH3), 2.40 (1H, m, 4′-H), 2.75 (1H, m, 4′-H), 3.88 (1H, m, 5′-H), 5.08 (1H, q, J 6.7, 3-H), 5.35 (1H, dd, J 12.9, 5.9, 6′-H), 5.76 (1H, m, 3′-H), 5.83 (1H, m, 1′-H), 6.17 (1H, m, 2′-H), 7.16–7.35 (5H, m, Ar-H), 7.37–7.49 (2 × 1H, 2 × m, 4-H, 6-H), 7.55 (1H, m, 5-H), 7.81 (1H, d, J 7.2, 7-H); m/z (%) 349 (M+
+ 1, 12), 318 (10), 302 (67), 199 (72), 155 (81), 148 (100).
(3aS*,4R*,7aS*,4′R*)-4-(4′-Isopropyl-2′-oxooxazolidin-3′-yl)-2-phenyl-2,3,3a,4,7,7a-hexahydro-1H-isoindole-1,3-dione 26.
A solution of (1′E)-N-(buta-1′,3′-dienyl)-4-isopropyloxazolidin-2-one (0.1 g, 0.55 mmol) and N-phenylmaleimide (96 mg, 0.55 mmol) in chloroform (10 ml) was stirred at room temperature for 12 h. Concentration followed by recrystallisation from diisopropyl ether–methanol gave the title compound (0.13 g, 67%) as white crystals, mp 220–221 °C (C20H22N2O4 requires C, 67.8; H, 5.7; N, 7.9%. Found C, 67.5; H, 5.8; N, 7.7%); νmax (KBr)/cm−1 2966, 2938, 2850, 1738, 1596; δH (500 MHz, CDCl3) 0.91 (3H, d, J 7.0, CH3),
0.99 (3H, d, J 6.8, CH3), 2.10 (1H, m, CH3CH), 2.29 (1H, m, 7-H), 2.93 (1H, dd, J 15.9, 5.7, 7-H), 3.34 (1H, t, J 7.9, 7a-H), 3.89 (1H, dd, J 5.7, 2.7, 4′-H), 3.98 (1H, dd, J 8.8, 7.3, 3a-H), 4.20 (1H, dd, J 8.2, 2.7, 5′-H), 4.43 (1H, dd, J 8.2, 5.7, 5′-H), 4.53 (1H, d, J 3.6, 4-H), 6.11–6.19 (2 × 1H, 2 × m, 5-H, 6-H), 7.22 (2H, d, J 7.8, Ar–H), 7.30–7.47 (3H, m, Ar-H); m/z (%) 354 (M+, 0.45), 310 (23), 226 (31), 79 (100).
(3aS,4R,7aS,4′R*)-(−)-4-(2′-Oxo-4′-phenyloxazolidin-3′-yl)-2-phenyl-2,3,3a,4,7,7a-hexahydro-1H-isoindole-1,3-dione 27.
A solution of (4R,1′E)-N-(buta-1′,3′-dienyl)-4-phenyloxazolidin-2-one (0.22 g, 1.0 mmol) and N-phenylmaleimide (0.19 g, 1.1 mmol) in chloroform (10 ml) was stirred at room temperature for 12 h. The solvent was removed and the solid recrystallised from diisopropyl ether–methanol to give the title compound (0.29 g, 75%) as white prisms, mp 195–197 °C; [α]D
=
−43.6 (c 0.6, CHCl3) (C23H20N2O4 requires C, 71.1; H, 5.2; N, 7.2%. Found C, 71.0; H, 5.2; N, 7.1%); νmax (KBr)/cm−1 2922, 2844, 1759, 1706; δH
(500 MHz, CDCl3) 2.20 (1H, m, 7-H), 2.86 (1H, ddd, J 15.8, 7.0, 1.8, 7-H), 3.30 (1H, ddd, J 9.3, 7.0, 1.7, 7a-H), 3.81 (1H, dd, J 9.3, 7.0, 3a-H), 4.12 (1H, dd, J 8.4, 3.4, 5′-H), 4.61 (1H, dq, J 7.0, 3.1, 4-H), 4.81 (1H, t, J 8.4, 4′-H), 5.02 (1H, dd, J 8.4, 3.4, 5′H), 5.77 (1H, dt, J 9.8, 3.2, 5-H), 5.89 (1H, m, 6-H), 7.23–7.47 (10H, m Ar-H); δC (125 MHz, CDCl3) 15.3, 24.0, 38.5, 42.0, 50.4, 59.7, 71.6, 126.0, 126.5, 128.7, 128.8, 129.3, 128.5, 131.8, 141.0 158.3, 176.5, 178.2; m/z (%) 388 (M+ 2.5), 344 (60), 227 (55), 215 (83), 170 (54), 162 (67), 104 (100), 91 (63), 80 (88), 32 (100).
(3aS,4R,7aS,4′R*)-(−)-6-Methyl-4-(2′-oxo-4′-phenyloxazolidin-3′-yl)-2-phenyl-2,3,3a,4,7,7a-hexahydro-1H-isoindole-1,3-dione 28.
A solution of (4R,1′E)-3-(3′-methylbuta-1′,3′-dienyl)-4-phenyloxazolidin-2-one (23 mg, 0.1 mmol) and N-phenylmaleimide (17 mg, 0.1 mmol) in chloroform (1 ml) was stirred at room temperature for 2 h. Removal of the solvent followed by crystallisation from diisopropyl ether–methanol gave the title compound (28 mg, 69%) as white crystals, mp 139–140 °C; [α]D
=
−34.5 (c 0.6, EtOH) (C24H22N2O4 requires 402.1579. Found M+ 402.1577); νmax (KBr)/cm−1 2967, 2924, 2844, 1739, 1707; δH
(500 MHz, CDCl3) 1.67 (3H, s, CH3), 2.23 (1H, m, 7-H), 2.69 (1H, dd, J 15.4, 1.9, 7-H), 3.28 (1H, m, 7a-H), 3.72 (1H, dd, J 8.9, 7.9, 3a-H), 4.11 (1H, dd, J 8.1, 3.6, 5′-H), 4.55 (1H, m, 4-H), 4.80 (1H, t, J 8.4, 4′-H), 4.97 (1H, dd, J 8.5, 3.6, 5′-H), 5.42 (1H, d, J 1.1, 5-H), 7.23–7.49 (10H, m, Ar-H); δC (125 MHz, CDCl3) 23.5, 28.9, 38.8, 41.8, 50.8, 59.7, 71.5, 118.8, 126.0, 126.4, 128.7, 128.8, 129.2, 129.3, 131.8, 138.1, 140.9, 158.3, 176.5, 178.1; m/z (%) 402 (M+, 2.8), 358 (12.2), 162 (80), 104 (100), 77 (47).
(1S,5R,6S,4′R)-(−)-6-Nitro-5-(2′-oxo-4′-phenyloxazolidin-3-yl)cyclohex-3-enecarboxylic acid ethyl ester 29.
A solution of (4R,1′E)-N-(buta-1′,3′-dienyl)-4-phenyloxazolidin-2-one (0.39 g, 1.8 mmol) and ethyl nitroacrylate (0.30 g, 2.1 mmol) in chloroform (15 ml) was stirred at room temperature for 2 h. Concentration, followed by trituration with ethanol gave a white solid which was recrystallised from ethanol to give the title compound (0.42 g, 65%) as white prisms, mp 135 °C; [α]D
=
−390.5 (c 1.9, CHCl3) (C18H20N2O6 requires C, 60.0; H, 5.6; N, 7.8%. Found C 60.0; H 5.7; N, 7.6%); νmax (KBr)/cm−1 3041, 2982, 2940, 2913, 1749, 1566, 1404, 1326, 1208,
1078, 709; δH (500 MHz, CDCl3) 1.28 (3H, t, J 7.1, CH3), 2.13 (1H, m, 2-H), 2.56 (1H, m, 2-H), 3.56 (1H, td, J 11.5, 6.0, 1-H), 4.13 (1H, dd, J 8.6, 2.8, 5′-H), 4.21 (2H, q, J 7.1, OCH2), 4.53 (1H, t, J 8.6, 4′-H), 4.87 (1H, dd, J 8.6, 2.8, 5′-H), 5.08 (1H, dd, J 11.5, 6.1, 6-H), 5.19 (1H, m, 5H), 5.23 (1H, m, 4-H), 5.48 (1H, m, 3-H), 7.23 (2H, m, Ar-H), 7.35 (3H, m, Ar-H); δC (125 MHz, CDCl3) 14.1, 27.7, 39.7, 49.1, 59.6. 61.8, 70.8, 84.6,122.6, 126.9, 128.2, 129.11, 129.16, 139.8, 157.7, 171.6; m/z (%) 360 (M+, 0.4), 240 (100), 196 (45), 120 (33), 104 (95), 77 (72), 29 (54).
Acknowledgement
We would like to thank EPSRC for grant GR/K10430 in support of this work.
References
- J. Barluenga, A. Suarez-Sobrino and L. A. Lopez, Aldrichimica Acta, 1999, 32, 4 Search PubMed.
- W. Oppolzer and W. Frostl, Helv. Chim. Acta, 1975, 58, 593 CrossRef CAS.
- W. Oppolzer and K. Keller, J. Am. Chem. Soc., 1971, 93, 3836 CrossRef CAS.
- W. Oppolzer and W. Frostl, Helv. Chim. Acta, 1975, 58, 590 CrossRef CAS.
- L. E. Overman, G. F. Taylor, K. N. Houl and L. N. Domelsmith, J. Am. Chem. Soc., 1978, 100, 3182 CrossRef CAS.
- L. E. Overman and P. J. Jessup, J. Am. Chem. Soc., 1978, 100, 5179 CrossRef CAS.
- L. E. Overman, R. L. Freerks, C. B. Petty, L. A. Clizbe, R. K. Ono, G. F. Taylor and P. J. Jessup, J. Am. Chem. Soc., 1981, 103, 2816 CrossRef CAS.
- L. E. Overman, D. Lesuisse and M. Hashimoto, J. Am. Chem. Soc., 1983, 105, 5373 CrossRef CAS.
- L. E. Overman and R. L. Freerks, J. Org. Chem., 1981, 46, 2833 CrossRef CAS.
- S. Masamune, L. A. Reed, J. T. Davis and W. Choy, J. Org. Chem., 1983, 48, 4441 CrossRef CAS.
- S. Blasco, M. C. Carreno, M. B. Cid, J. L. G. Ruano and M. R. Martin, Tetrahedron: Asymmetry, 1999, 10, 3473 CrossRef CAS.
- C. A. Zezza and M. B. Smith, J. Org. Chem., 1988, 53, 1161 CrossRef CAS.
- R. F. Menezes, C. A. Zezza, J. Sheu and M. B. Smith, Tetrahedron Lett., 1989, 30, 3295 CrossRef CAS.
- J. B. Behr, A. Defoin, J. Pires, J. Streith, L. Macko and M. Zehnder, Tetrahedron, 1996, 52, 3283 CrossRef CAS.
- A. Defoin, J. Pires, I. Tissot, T. Tschamber, D. Bur, M. Zehnder and J. Streith, Tetrahedron: Asymmetry, 1991, 2, 1209 CrossRef CAS.
- J. M. Janey, T. Iwama, S.
A. Kozmin and V. H. Rawal, J. Org. Chem., 2000, 65, 9059 CrossRef CAS.
- Y. Huang and V. H. Rawal, Org. Lett., 2000, 2, 3321 CrossRef CAS.
- S. A. Kozmin, M. T. Green and V. H. Rawal, J. Org. Chem., 1999, 64, 8045 CrossRef CAS.
- S. A. Kozmin and V. H. Rawal, J. Am. Chem. Soc., 1999, 121, 9562 CrossRef CAS.
- S. A. Kozmin, J. M. Janey and V. H. Rawal, J. Org. Chem., 1999, 64, 3039 CrossRef CAS.
- S. A. Kozmin and V. H. Rawal, J. Org. Chem., 1997, 62, 5252 CrossRef CAS.
- S. A. Kozmin and V. H. Rawal, J. Am. Chem. Soc., 1997, 119, 7165 CrossRef.
- D. A. Evans, S. J. Miller, T. Lectka and P. von Matt, J. Am. Chem. Soc., 1999, 121, 7559 CrossRef CAS.
- Y. Huang, T. Iwama and V. H. Rawal, J. Am. Chem. Soc., 2000, 122, 7843 CrossRef CAS.
- A. I. Khalaf, S. Linaza, A. R. Pitt, W. H. Stimson and C. J. Suckling, Tetrahedron, 2000, 56, 489 CrossRef CAS.
- J. P. Murphy, M. Nieuwenhuyzen, K. Reynolds, P. K. S. Sarma and P. J. Stevenson, Tetrahedron Lett., 1995, 36, 9533 CrossRef CAS.
- R. W. Hoffmann, Chem. Rev., 1989, 89, 1841 CrossRef CAS.
- W. Oppolzer, M. Wills, M. J. Kelly, M. Signer and J. Blagg, Tetrahedron Lett., 1990, 31, 5015 CrossRef CAS.
- A. Kumar, Chem. Rev., 2001, 101, 1 CrossRef CAS.
- P. A. Grieco, Aldrichimica Acta, 1991, 24, 59 Search PubMed.
- M. Ayerbe and F. P. Cossio, Tetrahedron Lett., 1995, 36, 4447 CrossRef CAS.
- A. Guy and L. Serva, Synlett, 1994, 647 CrossRef CAS.
- S. M. Allin, C. J. Northfield, M. I. Page and A. M. Z. Slawin, J. Chem. Soc., Perkin Trans. 1, 2000, 11, 1715 RSC.
- J. Montgomery, G. M. Wieber and L. S. Hegedus, J. Am. Chem. Soc., 1990, 112, 6255 CrossRef CAS.
- A. Guillam, L. Toupet and J. Maddaluno, J. Org. Chem., 1998, 63, 5110 CrossRef CAS.
- R. Huisgen, K. Herbig, A. Siegl and Huber, Chem. Ber., 1966, 2526 Search PubMed.
- P. C. B. Page, S. A. Harkin, A. P. Marchington and M. B. Vanniel, Tetrahedron, 1989, 45, 3819 CrossRef CAS.
- C. H. McMullen and J. C. M. Stirling, J. Chem. Soc. B, 1966, 1217 RSC.
- A. F. Barrero, J. F. Q. Del Moral, J. M. Cuerva, E. Cabrera and D. Jimenez-Gonzalez, Tetrahedron Lett., 2000, 41, 5203 CrossRef CAS.
- D. F. Oliveira, P. C. M. L. Miranda and C. R. D. Correia, J. Org. Chem., 1999, 64, 6646 CrossRef CAS.
- D. A. Evans and E. B. Sjogren, Tetrahedron Lett., 1985, 26, 3783 CrossRef CAS.
- F. Jeanjean, G. Fournet, D. Le Bars and J. Gore, Eur. J. Org. Chem., 2000, 1297 CrossRef CAS.
- T. Bach, J. Schroder, T. Brandl, J. Hecht and K. Harms, Tetrahedron, 1998, 54, 4507 CrossRef CAS.
- A. B. Bueno and L. S. Hegedus, J. Org. Chem., 1998, 63, 684 CrossRef CAS.
- J. W. Fisher, J. M. Dunigan, L. D. Hatfield, R. C. Hoying, J. E. Ray and K. L. Thomas, Tetrahedron Lett., 1993, 34, 4755 CrossRef CAS.
- SAINT-NT, Brüker AXS Madison, Wisconsin, 1998.
- XSCANS, J. Fait, Brüker AXS Madison, Wisconsin, 1993.
-
G. M. Sheldrick, University of Göttingen, Göttingen, Germany, 1998.
|
| This journal is © The Royal Society of Chemistry 2002 |
Click here to see how this site uses Cookies. View our privacy policy here.