Diastereocontrolled synthesis of hydroxylated lactams
Mark. D. Andrewsa, Andrew G. Brewsterb and Mark G. Moloney*a
aThe Department of Chemistry, The Dyson Perrins Laboratory, The University of Oxford, South Parks Road, Oxford, UK OX1 3QY. E-mail: mark.maloney@chem.ox.ac.uk
bZeneca Pharmaceuticals, Mereside, Alderley Park, Macclesfield, UK SK10 4TG
First published on 5th December 2001
Abstract
Highly diastereoselective reductions and organometallic additions in bicyclic lactams have been observed, which appear to result either from a stereoelectronic interaction of the pyramidalised nitrogen lone pair or from steric interactions in the bicyclic system. These products can be readily deprotected to give hydroxylated lactams.
Highly substituted lactam rings possess diverse biological activity, and much recent attention has focussed on excitatory amino acid chemistry, particularly as a result of the drive to better understand CNS function in mammalian systems.1 It has recently also been shown that densely functionalised pyrrolidinones, such as lactacystin,2 exhibit potent and selective activity in proteasome inactivation; there has therefore been considerable interest in the rapid construction of the pyrrolidinone framework,3 since such compounds may find application in the development of selective therapeutic agents for important parasitic infections.4,5
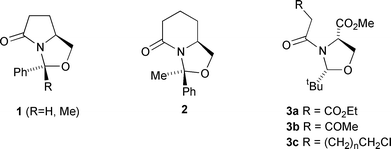
Our interest in this area has been concerned with the application of bicyclic lactams for the synthesis of diversely substituted lactam rings. Two lactam templates 1 and 2, derived from pyroglutamic or pyroadipic acids respectively, have been used to access a variety of kainoid analogues as well as novel aminolactams, and make use of the inherent facial selectivity which comes from the bicyclic system.6–9 Another template 3, derived from serine,10 has also been found to be applicable for the preparation of diversely functionalised lactams of well-defined stereochemistry. In this series, ring closure of 3a–c by Dieckmann, Aldol or intramolecular cyclisation reactions respectively led to functionalised lactam systems which possess a quaternary α-amino centre with high diastereo- and enantiocontrol.11–14 We wished to further develop this chemistry to permit the introduction of hydroxy functionality around the ring periphery, and describe here that highly diastereocontrolled ring modifications using hydride or organometallic nucleophiles in bicyclic lactams are possible using steric, chelation or stereoelectronic control.
Results and discussion
Starting material synthesis
Lactams 5a,b were prepared by our previously reported methodology (Scheme 1);11,12 oxazolidine 4, itself readily obtained from serine and pivaldehyde in 90% yield, was acylated with the required β-dicarbonyl unit and then cyclised to the tetramic acid by treatment with potassium tert-butoxide in tert-butyl alcohol.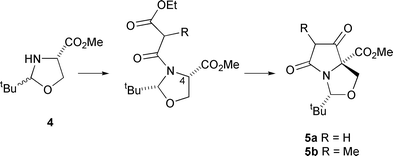 | ||
| Scheme 1 | ||
Ketone reductions
Our initial investigations were concerned with selective reductions in the bicyclic lactams 5a,b; for this purpose, sodium triacetoxyborohydride appeared to be the reagent of choice, since Poncet and co-workers had earlier reported that reduction of the related bicyclic tetramic acid 6 with this reagent generated in situ from sodium borohydride and acetic acid in dichloromethane gave the alcohols 7a,b in the ratio 95 ∶ 5 (Scheme 2).15 This corresponds to preferential attack of the ketone carbonyl by borohydride from the sterically more hindered endo-face of the bicyclic system, anti to the nitrogen lone pair, and from a study of several substrates, the authors concluded that this was a general phenomenon.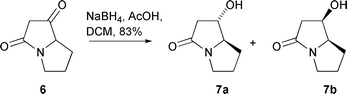 | ||
| Scheme 2 | ||
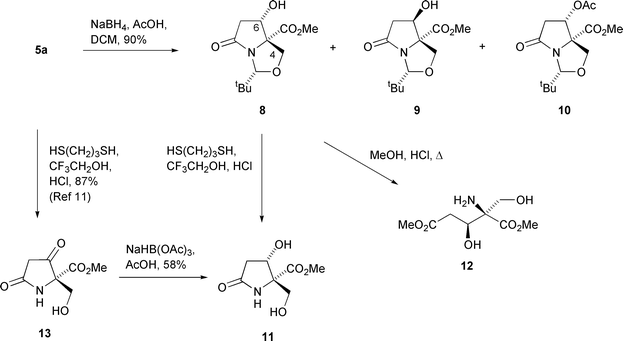
Application of this method to the reduction of dicarbonyl compound 5a gave a very similar result. Thus, reduction with sodium borohydride in a 1 ∶ 9 mixture of acetic acid and dichloromethane gave a 63 ∶ 1 mixture of the epimeric alcohols 8 and 9, along with 0.1% of the acetate 10 (Scheme 3). That the major product was the alcohol 8, with the indicated relative stereochemistry, was initially assumed on the basis of the results obtained by Poncet, but this was later confirmed by single crystal X-ray analysis of a derivative (vide infra). Recrystallisation from chloroform–petrol gave the major epimer 8 in a yield of 81%. The relative stereochemistry of compound 10 was assumed to be the same as the major reduction product 8, from which it was presumably formed under the reaction conditions.
 | ||
| Scheme 3 | ||
Deprotection of lactam 8 to diol 11 was achieved in quantitative yield in a mixture of propane-1,3-dithiol and acidic trifluoroethanol.16 This synthesis of ester 11, which was without recourse to column chromatography, was achieved in an excellent total yield of 42% from L-serine. Alternatively, both the oxazolidine and lactam rings of alcohol 8 could be cleaved by heating in methanolic HCl at reflux; after removal of solvent in vacuo and purification by column chromatography, the glutamate ester 12, contaminated with 8% of the diol 11, was obtained. Diol 11 could also be synthesised, in lower overall yield, by reversing the order of the last two steps in the synthesis above. Thus, deprotection of dicarbonyl 5a as described above gave the unstable keto alcohol 13 in 87% yield.11 Treatment of the crude product with sodium triacetoxyborohydride in acetic acid then led to the diol 11 in 58% yield.
Confirmation of the stereochemical assignment above was possible by successful derivatisation to a crystalline compound. Alcohol 8 was protected in quantitative yield with benzoyl chloride and pyridine to give the benzoate ester 14 (Scheme 4) and deprotection with propane-1,3-dithiol in acidic trifluoroethanol gave alcohol 15a in 97% yield. Attempted N-Boc protection in fact gave the crystalline carbonate 15b in 92% yield, whose relative stereochemistry was confirmed by single crystal X-ray structural analysis.17 This important result confirmed the earlier tentative stereochemical assignments of compounds 8, 9 and 10. O,N-Diprotection, using triethylamine as a base, with a catalytic amount of DMAP as a nucleophilic catalyst, in fact gave the elimination product 16 in 58% yield along with a 31% yield of the carbonate 15b.
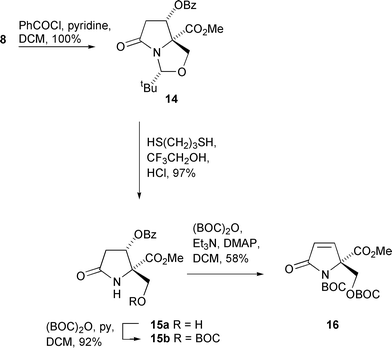 | ||
| Scheme 4 | ||
In view of the success of these reductions, it was of interest to examine the diastereocontrol of more elaborate systems of type 5b.† Reduction of dicarbonyl 5b with sodium borohydride in acetic acid and dichloromethane as described above gave, in addition to 29% of unreacted starting material, three diastereomeric alcohols 17a, 17b and 17c, which were obtained, after careful purification, in yields of 32, 16 and 4% respectively (Scheme 5). Interestingly, it was found that this reduction needed to be carried out in the absence of oxygen to prevent autoxidation, otherwise the diol product 18 was obtained in low yield.11 The relative stereochemistries of these three alcohols were established by a series of NOE experiments (Fig. 1). For 17a and 17b, a sequence of irradiations easily gave the relative stereochemistry around the bicyclic ring system, but for the minor product 17c only one enhancement gave any exact information and the overall stereochemistry was therefore assumed from the earlier assignments of isomers 17a,b. Hydride addition, predominantly from the endo-face, which is anti to the nitrogen lone pair gives the major products 17a,b. The low overall yield for the reduction can probably be explained by steric hindrance from the C(7) methyl group.
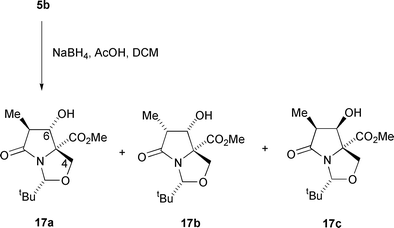 | ||
| Scheme 5 | ||
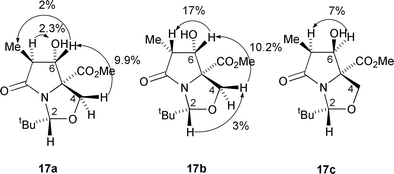 | ||
| Fig. 1 | ||

In order to gain some information concerning the stability of these reduction products, epimerisation studies of the epimeric alcohols 17a or 17b were undertaken with sodium methoxide in methanol. For the former, no change was observed on stirring with base but lactam 17b gave almost complete epimerisation to its C(7) epimer 17a (de 94%), clearly indicating that alcohol 17a is the thermodynamically more stable. Epimerisation at C(6) of the alcohols 17a and 17b was also attempted under the Mitsunobu conditions,18 but treatment with benzoic acid, triphenylphosphine and DEAD in THF gave none of the desired products, with the only compound isolated being the alkene 19. Alcohol 17a gave a 36% yield of this elimination product with 53% of starting material being recovered, while alcohol 17b gave alkene 19 in 33% yield along with 38% recovered starting material.
In an attempt to improve the reduction of dicarbonyl compound 5b, it was decided to change the order of the steps in the synthesis by cleaving the oxazolidine ring before attempting the reduction of the ketone. Accordingly, dicarbonyl compound 5b was converted to the unstable keto alcohol 20 as reported previously (Scheme 6).11 Reaction with sodium triacetoxyborohydride then gave diol 21 as an inseparable 3 ∶ 2 mixture of two diastereomers in 51% yield. It was assumed that these diols were epimeric at C(4) rather than C(3) because of the excellent diastereocontrol exhibited previously in the sodium triacetoxyborohydride reduction of lactam 13.
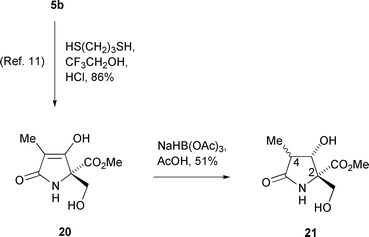 | ||
| Scheme 6 | ||
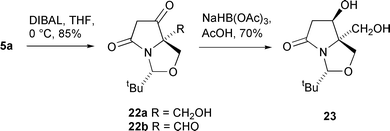
In view of the good to high levels of diastereoselectivity exhibited in ring carbonyl reductions, it was of interest to examine manipulation of the pyrrolidinone ring side-chains. Reduction of the C(5) ester function of 5a proved to be possible with 4 equivalents of a solution of DIBAL-H in hexane in THF at 0 °C, giving the crude alcohol 22a in 85% yield (Scheme 7), although this compound was found to be unstable to chromatography. Attempts to oxidise this material to the aldehyde 22b with TPAP or PCC were unsuccessful, and although evidence for successful Swern oxidation could be obtained by 1H NMR analysis of the crude product, this material also proved to be unstable to isolation. Further reduction of 22a with sodium triacetoxyborohydride in acetic acid gave diol 23 as a single diastereomer in 70% yield; the stereochemistry of this product was assumed to be as shown because of the presumed intramolecular chelated delivery of the hydride in the triacetoxyborohydride reduction, forcing hydride attack from the Si face of the ketone of 22a.
 | ||
| Scheme 7 | ||
In several of the above compounds, it was noted that consistent trends were observed in the 1H NMR spectra which facilitated assignment of stereochemistry. Thus, for the C(6)Hendo compounds 8, 10, 17a and 17b, the difference in the C(4)Hendo and C(4)Hexo chemical shift values were typically 1.1–1.4 ppm, and were much larger than in the C(6)Hexo compounds 9, 17c and 23, for which the difference was only 0.3 ppm. Conversely, the difference in C(7)Hendo and C(7)Hexo chemical shift values for the C(6)Hendo compounds 8 and 10 were typically only 0.4 ppm, significantly lower than the 0.9 ppm observed for the C(6)Hexo compounds 9, 17c and 23. Similar trends in NMR data in functionalised pyrrolidinones have been ascribed to steric compression of the different proton nuclei around the periphery of the heterocyclic system.19
Organometallic additions
In view of the success of the diastereoselective reductions, it was also of interest to examine the possibility of introducing alcohol functionality using organometallic additions. Kikkawa and Yorifuji reported that, in the presence of triethylamine, aliphatic Grignard reagents react with esters to form the corresponding ketones in good yield.20 Unfortunately, lactams 5a,b were unreactive with isopropylmagnesium bromide, even on heating in THF at reflux, but with the more reactive alkyllithium species (5 equivalents in THF–petrol at −78 °C21,22), ester 5a gave an inseparable 3 ∶ 1 mixture of ketone 24a and the secondary alcohol 25a in a total yield of 85% (Scheme 8). No tertiary alcohol due to reaction of ketone 24a with another equivalent of isopropyllithium was observed in this reaction, presumably since this is a particularly hindered ketone. The ketone 24a–alcohol 25a product could be entirely converted to the alcohol product 25a using lithium in ammonia–ethanol in 70% yield for the two steps from ester 5a. Similarly, ester 5b was reacted with excess isopropyllithium to give a partially separable 7 ∶ 1 mixture of ketone 24b and secondary alcohol 25b. The formation of the secondary alcohols 25a,b was due to reduction of the ketones 24a,b by excess lithium metal in the reaction.23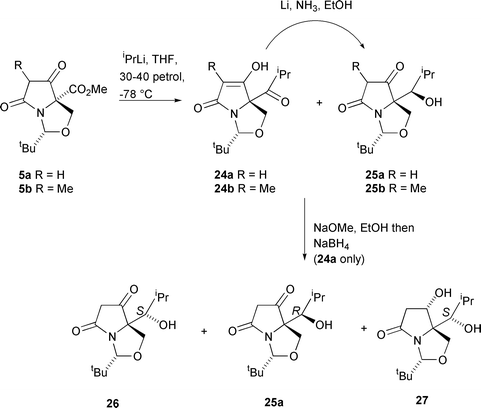 | ||
| Scheme 8 | ||
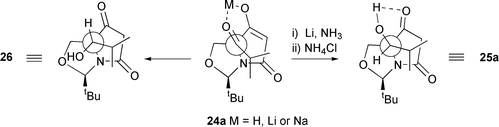
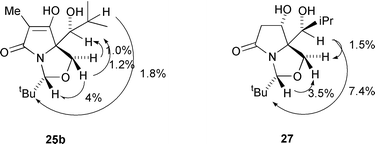
Reduction of the ketone 24a–alcohol 25a mixture with alkaline sodium borohydride in ethanol (in which the β-dicarbonyl enolate was formed, thereby protecting the ketone from reduction) gave 3 products: (S)-alcohol 26 was obtained in 36% yield, along with 19% of (R)-alcohol 25a, present as a contaminant in the starting material, and a 20% yield of the over-reduction product, diol 27. The yields of these products are quoted for the two steps from ester 5a, and their stereochemistry was assigned by a consideration of their IR spectral data. Thus, the product 25a, also obtained by dissolving metal reduction (Li, NH3, EtOH) of ketone 24a as described above, exhibited a broad peak from 3600 to 2200 cm−1 and peaks at 1680, 1650 and 1610 cm−1, consistent with the presence of an intramolecular hydrogen bond. For the epimer 26, the IR spectrum showed no evidence for such intramolecular hydrogen bonding, with the OH signal occurring at 3600–3100 cm−1 and the carbonyl signal occurring at 1770 and 1710 cm−1. These observations are consistent with reduction of the H-bonded ketone 24a (or its metal chelated enolate, Fig. 2) which in the case of dissolving metal reduction, leads to the thermodynamically more stable H-bonded product 25a in which steric interactions between the iBu and iPr groups are minimised. On the other hand, reduction of 24a by NaBH4 occurs by attack of hydride at the least hindered face, as indicated, to give the product 26 in which intramolecular hydrogen bonding is not possible, without causing unfavourable steric interactions between the iBu and iPr groups. That compounds 26 and 27 possessed the same side-chain stereochemistry (S) was established by the conversion of ketone 26 to alcohol 27 by catalytic hydrogenation with platinum oxide in ethyl acetate at 5 atm; under these conditions, the reaction was slow and gave only a low yield (25%) of product 27, along with recovered starting material. The stereochemistry of 27 was in part determined by NOE (Fig. 3), which established the side-chain stereochemistry, and in part by use of the 1H NMR patterns discussed above; thus, the Δδ of each of the H(4) signals, at 1.0 ppm, was consistent with the exo orientation of the C(6) hydroxy as indicated. Such a stereochemistry would have required hydrogen attack anti to the nitrogen lone pair, which, as noted above, appears to be the kinetically preferred mode of attack, and in any case the alternative exo-face reduction is clearly blocked by the bulky iPr group.
 | ||
| Fig. 2 | ||
 | ||
| Fig. 3 | ||
Reduction of ketone 24b was similarly achieved; treatment with sodium methoxide in ethanol followed by addition of sodium borohydride under nitrogen led to the (S)-alcohol 28 in 91% yield (Scheme 9). This reaction was performed under nitrogen because both product alcohols 28 and 25b were found to be extremely sensitive to autoxidation. On the other hand, dissolving metal reduction of ketone 24b with lithium in liquid ammonia gave only the (R)-alcohol 25b in 89% yield. Use of sodium in liquid ammonia, however, led to a mixture of the (R)- and (S)-alcohols, the (R)-alcohol 25b predominating, in a combined yield of 57%. The stereochemistries of the (R)- and (S)-alcohols 25b and 28 were assigned by analogy with those of alcohols 25a and 26.
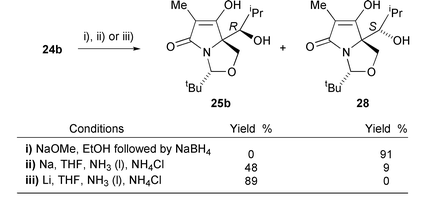 | ||
| Scheme 9 | ||
Again, useful patterns in the 1H NMR spectra were observed: the appearance of the methine proton of the isopropyl side-chain in the 1H NMR spectra of these compounds proved to be diagnostic of the alcohol stereochemistry. For those compounds assigned the (S)-stereochemistry, this methine proton appeared as an octet, with the coupling constants to the isopropyl methyl groups and the proton at the alcohol centre all being approximately 6.5 or 7.0 Hz. For those compounds assigned the (R)-stereochemistry, however, this methine proton appeared as a doublet of septets or a septet, the coupling constant to the proton at the alcohol centre being 2.0 Hz or less.
As noted above, alcohols 25b and 28 were found to be unusually sensitive to aerial oxidation, and attempted recrystallisation of alcohol 28 from chloroform led to a decomposition owing to this instability. Purification of this decomposition mixture by column chromatography enabled the isolation of diol 29 in 7% yield. That the oxidation product was indeed the diol 29 was confirmed by mass spectrometry, both chemical ionisation and positive electrospray spectra showing peaks at 317 and 300 Da corresponding to M + NH4+ and M + H+ ions respectively. Storage of alcohol 25b in deuterochloroform solution for 20 days led to relatively clean oxidation to give a single product as shown by 1H NMR spectroscopy. Purification by column chromatography gave unchanged starting material in 50% recovery and a 30% yield of the peroxide 30. That this compound was the peroxide and not the diol analogous to compound 29 was established from a positive starch iodide test. This was supported by mass spectrometry, and the positive electrospray spectrum of compound 30 clearly showed a peak at 316 Da indicating that the peroxide was indeed the product formed.
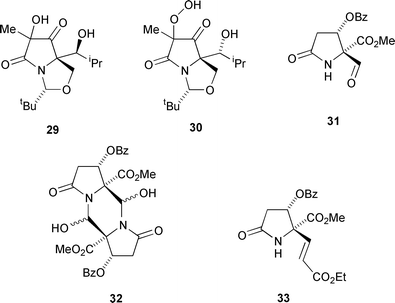
In view of the successful modification of the C(5) ester function of 5 by organometallic addition, it was of interest to investigate the reactions of the corresponding aldehyde. However, attempted oxidation of alcohol 15a to the aldehyde 31 using Swern conditions gave not this product, but the corresponding dimer 32 in 42% yield; disappointingly, although this compound gave an acceptable yield (56%) of product 33 (along with 5% of the Z-isomer) by reaction with the stabilised Wittig reagent, non-stabilised ylides and Grignards, with or without Lewis acid activation, gave none of the desired products.
Conclusion
We have shown that hydroxylated pyrrolidinones are available by reduction or organometallic addition to protected tetramic acid derivatives, which in the best cases proceed with excellent diastereocontrol. Deprotection permits rapid access to highly functionalised pyrrolidinones.Experimental
Lactams 5a,b were prepared by our previously reported methodology.11(2R,5R,6S)- and (2R,5R,6R)-2-tert-Butyl-6-hydroxy-5-methoxycarbonyl-8-oxo-3-oxa-1-azabicyclo[3.3.0]octane 8 and 9 and (2R,5R,6S)-6-acetoxy-2-tert-butyl-5-methoxycarbonyl-8-oxo-3-oxa-1-azabicyclo[3.3.0]octane 10
To a solution of lactam 5a (9.0 g, 35.3 mmol) in 10% AcOH in DCM (175 ml) at 0 °C was added, portionwise over 15 min, NaBH4 (2.93 g, 77.6 mmol). The mixture was stirred at 0 °C for a further 15 min, at room temperature for 1.5 h and the solvent was then removed in vacuo. The residue was partitioned between EtOAc (250 ml) and sat. NaHCO3 (aq) (250 ml) and the aqueous layer was re-extracted with EtOAc (2 × 100 ml). The aqueous layer was then acidified with 1M HCl and extracted with EtOAc (250 ml), the EtOAc layer being washed with brine (200 ml), dried (MgSO4) and evaporated in vacuo to give recovered starting material 5a (0.25 g, 2.8%). The neutral organic extracts were washed with brine (300 ml), dried (MgSO4) and evaporated in vacuo to give a crude mixture of epimeric alcohols 8 and 9 (ratio 63 ∶ 1). Recrystallisation from CHCl3–hexane gave alcohol 8 (7.36 g, 81%), contaminated with traces (<1.5%) of alcohol 9, as a colourless crystalline solid; mp 129–130.5 °C; Rf = 0.07 (EtOAc–DCM, 1 ∶ 3); [α]20D = +72.4 (c = 0.98 in CHCl3); found: C, 55.9; H, 7.3; N, 5.3; C12H19NO5 requires: C, 56.0; H, 7.4; N, 5.5%; νmax (Nujol) 3325 (br m), 1740 (s), 1690 (s) and 1680 (s) cm−1; δH (200 MHz, CDCl3) 0.87 (9H, s, C(CH3)3), 2.83 (1H, dd, J = 16.0 Hz, J′ = 8.0 Hz, CHH′CH), 3.05 (1H, br s, OH), 3.16 (1H, dd, J = 16.0 Hz, J′ = 10.5 Hz, CHH′CH), 3.57 (1H, d, J = 8.5 Hz, C(4)HH′), 3.83 (3H, s, CO2CH3), 4.57 (1H, m, CHH′CH), 4.87 (1H, s, tBuCH) and 4.89 (1H, d, J = 8.5 Hz, C(4)HH′); δC (50.3 MHz, CDCl3) 24.7 (C(CH3)3), 36.0 (C(CH3)3), 43.0 (C(7)H2), 52.6 (CO2CH3), 75.6 (C(4)H2 and C(6)H), 77.5 (C(5)), 96.2 (tBuCH) and 170.7 and 175.2 (carbonyls); m/z (CI) 258 (M + H+, 100%) and 200 (M + H+ − tBuH, 48).Purification of the residue by careful column chromatography (EtOAc–petrol, 2 ∶ 3) gave alcohols 8 (0.75 g, 8.3%) and 9 (0.04 g, 0.4%) as a crystalline solid; mp 104–105.5 °C; Rf = 0.12 (EtOAc–DCM, 1 ∶ 3); [α]25D = +29.4 (c = 0.35 in CHCl3); found: C, 56.03; H, 7.68; N, 5.40; C12H19NO5 requires: C, 56.02; H, 7.44; N, 5.45%; νmax (CHCl3) 3605 (w), 3500–3100 (br w), 2960 (m), 1740 (m), 1720 (s) and 1085 (s) cm−1; δH (200 MHz, CDCl3) 0.89 (9H, s, C(CH3)3), 2.46 (1H, d, J = 17.0 Hz, CHH′CH), 3.38 (1H, dd, J = 17.0 Hz, J′ = 6.0 Hz, CHH′CH), 3.81 (3H, s, CO2CH3), 4.24 (1H, d, J = 9.0 Hz, C(4)HH′), 4.41 (1H, t, J = 6.0 Hz, CHH′CH), 4.58 (1H, d, J = 9.0 Hz, C(4)HH′) and 4.92 (1H, s, tBuCH); δC (125.7 MHz, CDCl3) 24.88 (C(CH3)3), 36.21 (C(CH3)3), 44.65 (C(7)H2), 52.92 (CO2CH3), 67.30 and 71.16 (C(4)H2 and C(6)H), 77.89 (C(5)), 96.38 (tBuCH) and 171.9 and 177.2 (CO); m/z (CI) 275 (M + NH4+, 9%), 258 (M + H+, 100) and 200 (25), along with acetate 10 (5.11) (0.01 g, 0.1%) as a colourless oil, Rf = 0.24 (EtOAc–petrol, 3 ∶ 7); νmax (CHCl3) 2960 (m), 1750 (s), 125 (s) and 1080 (m) cm−1; δH (200 MHz, CDCl3) 0.86 (9H, s, C(CH3)3), 2.02 (3H, s, CH3CO2), 2.80 (1H, dd, J = 15.5 Hz, J′ = 8.0 Hz, CHH′CH), 3.27 (1H, dd, J = 15.5 Hz, J′ = 11.0 Hz, CHH′CH), 3.72 (1H, d, J = 9.5 Hz, C(4)HH′), 3.79 (3H, s, CO2CH3), 4.82 (1H, d, J = 9.5 Hz, C(4)HH′), 4.86 (1H, s, tBuCH) and 5.22 (1H, dd, J = 11.0 Hz, J′ = 8.0 Hz, CHH′CH); δC (125.7 MHz, CDCl3) 20.41 (CH3CO2), 24.76 (C(CH3)3), 36.06 (C(CH3)3), 40.00 (C(7)H2), 52.73 (CO2CH3), 73.39 and 73.91 (C(4)H2, C(5) and C(6)H), 95.99 (tBuCH) and 169.5, 170.0 and 173.1 (CO); m/z (GCMS) 300 (M + H+, 2%), 240 (100) and 182 (70).
(2R,3S)-3-Hydroxy-2-hydroxymethyl-2-methoxycarbonylpyrrolidin-5-one 11
To a solution of alcohol 8 (1.0 g, 3.89 mmol) in acidic 2,2,2-trifluoroethanol (2% w/v HCl) (15 ml) was added propane-1,3-dithiol (0.4 ml, 0.43 g, 4.0 mmol). The mixture was stirred at room temperature for 5 h and silica (5 ml) was added before the solvent was removed in vacuo. The silica loaded material was placed on top of a silica filled (50 ml) sinter funnel and eluted with EtOAc–hexane 1 ∶ 1 (200 ml) followed by MeOH–EtOAc, 1 ∶ 4 (200 ml) to give diol 11 (0.73 g, 99%) as an off-white, hygroscopic crystalline solid; mp 45–47 °C; Rf = 0.09 (MeOH–EtOAc, 1 ∶ 9); [α]25D = +16.9 (c = 1.13 in MeOH); found: C, 44.1; H, 6.1; N, 7.4; C7H11NO5 requires: C, 44.4; H, 5.9; N, 7.4%; νmax (Nujol) 3340 (br s), 3290 (br s), 1735 (s) and 1685 (s) cm−1; δH (200 MHz, CD3OD) 2.27 (1H, dd, J = 17.0 Hz, J′ = 3.5 Hz, C(4)HH′), 2.73 (1H, dd, J = 17.0 Hz, J′ = 7.0 Hz, C(4)HH′), 3.71 (1H, d, J = 11.0 Hz, CHH′OH), 3.76 (3H, s, CO2CH3), 3.91 (1H, d, J = 11.0 Hz, CHH′OH) and 4.36 (1H, dd, J = 7.0 Hz, J′ = 3.5 Hz, C(3)HOH); δC (50.3 MHz, CD3OD) 40.9 (C(4)H2), 52.8 (CO2CH3), 65.6 (CH2OH), 71.2 (C(3)H), 75.5 (C(2)) and 171.9 and 178.5 (CO); m/z (CI) 207 (M + NH4+, 6%), 190 (M + H+, 100) and 130 (8).(2R,3S)-3-Hydroxy-2-hydroxymethylglutamic acid dimethyl ester 12
A solution of alcohol 8 (200 mg, 0.78 mmol) in methanolic HCl (5% w/v HCl) (10 ml) was heated at reflux for 7.5 h. After cooling to room temperature the solvent was removed in vacuo and the residue was purified by column chromatography (MeOH–EtOAc, 1 ∶ 9) giving glutamate 12 as a white foam contaminated with 8% of pyroglutamate 11 (170 mg); Rf = 0.17 (MeOH–EtOAc, 1 ∶ 9); νmax (Nujol) 3350 (br s), 1740 (s), 1440 (m) and 1250 (m) cm−1; δH (200 MHz, CD3OD) 2.49 (1H, dd, J = 16.0 Hz, J′ = 10.0 Hz, C(4)HH′), 2.71 (1H, dd, J = 16.0 Hz, J′ = 3.5 Hz, C(4)HH′), 3.71 (3H, s, CO2CH3), 3.72 (1H, d, J = 11.5 Hz, CHH′OH), 3.85 (3H, s, CO2CH3), 3.96 (1H, d, J = 11.5 Hz, CHH′OH) and 4.52 (1H, dd, J = 10.0 Hz, J′ = 3.5 Hz, C(3)H); δC (125.7 MHz, CD3OD) 37.99 (CH2CO2Me), 52.46 and 53.93 (2 × CO2CH3), 62.18 (CH2OH), 68.75 (CHOH), 70.21 (CNH2) and 170.7 and 172.6 (ester CO); m/z (CI) 222 (M + H+, 100%) and 190 (17).Alcohol 11 from lactam 13 by sodium triacetoxyborohydride reduction
To AcOH (3 ml) cooled in a water bath was added portionwise NaBH4 (38 mg, 1 mmol) and the mixture was stirred until H2 evolution ceased (about 10 min). A solution of keto alcohol 13 (90 mg, 0.48 mmol) in AcOH (2 ml) was then added dropwise and the mixture was stirred at room temperature for 5.5 h. Solvent was removed in vacuo and the residue was purified by column chromatography (EtOAc increasing polarity to MeOH–EtOAc, 1 ∶ 4) to give diol 11 (52 mg, 58%) as a hygroscopic, off-white crystalline solid. Data as described above.(2R,5R,6S)-6-Benzoyloxy-2-(tert-butyl)-5-methoxycarbonyl-8-oxo-3-oxa-1-azabicyclo[3.3.0]octane 14
To a solution of alcohol 8 (1.0 g, 3.89 mmol) and pyridine (1 ml, 0.72 g, 9.13 mmol) in DCM (25 ml) was added dropwise benzoyl chloride (1.0 ml, 1.22 g, 8.6 mmol). The mixture was heated at reflux for 24 h, allowed to cool to room temperature then partitioned between DCM (30 ml) and NH4Cl (aq) (40 ml). The organic layer was washed with 5% aq NaHCO3 (40 ml), water (40 ml) and brine (40 ml), dried (MgSO4) and evaporated in vacuo to give a quantitative yield of the title compound 14 as colourless needles contaminated with benzoic acid. An analytical sample was prepared by column chromatography (EtOAc–petrol, 1 ∶ 19 increasing polarity to EtOAc–petrol, 1 ∶ 4) (93% recovery); mp 174.5–175.5 °C; Rf = 0.56 (EtOAc–petrol, 3 ∶ 7); [α]25D = +7.2 (c = 0.61 in CHCl3); found: C, 63.23; H, 5.97; N, 3.76; C19H23NO6 requires: C, 63.14; H, 6.42; N, 3.88%; νmax (CHCl3) 2960 (m), 2870 (w), 1745 (m), 1725 (s) and 1600 (w) cm−1; δH (200 MHz, CDCl3) 0.90 (9H, s, C(CH3)3), 2.99 (1H, dd, J = 15.5 Hz, J′ = 8.0 Hz, C(7)HH′), 3.46 (1H, dd, J = 15.5 Hz, J′ = 11.0 Hz, C(7)HH′), 3.69 (3H, s, CO2CH3), 3.85 (1H, d, J = 9.0 Hz, C(4)HH′), 4.94 (1H, s, tBuCH), 4.95 (1H, d, J = 9.0 Hz, C(4)HH′), 5.50 (1H, dd, J = 11.0 Hz, J′ = 8.0 Hz, C(6)H), 7.43–7.51 (2H, m, ArH), 7.58–7.66 (1H, m, ArH) and 7.98 (2H, d, J = 7.0 Hz, ArH); δC (50.3 MHz, CDCl3) 24.6 (C(CH3)3), 35.8 (C(CH3)3), 40.1 (C(7)H2), 52.7 (CO2CH3), 73.7, 73.8 and 75.7 (C(4)H2, C(5) and C(6)H), 96.0 (tBuCH), 128.5, 128.8, 129.7 and 133.9 (aromatic) and 165.7, 169.6 and 173.6 (CO); m/z (CI) 379 (M + NH4+, 5%), 362 (M + H+, 100) and 304 (24).(2R,3S)-3-Benzoyloxy-2-hydroxymethyl-2-methoxycarbonylpyrrolidin-5-one 15a
To a solution of crude bicycle 14 (400 mg, 1.11 mmol) in acidic trifluoroethanol (1.5% w/v HCl) (15 ml) was added propane-1,3-dithiol (0.12 ml, 129 mg, 1.20 mmol). The mixture was stirred at room temperature for 22 h and solvent was then removed in vacuo at room temperature. Purification by column chromatography (EtOAc increasing polarity to MeOH–EtOAc, 1 ∶ 9) gave amido alcohol 15a (315 mg, 97%) as a crystalline solid; mp 138–139.5 °C; Rf = 0.29 (EtOAc); [α]25D = +61.9 (c = 1.11 in CHCl3); found: C, 57.21; H, 4.93; N, 4.57; C14H15NO6 requires: C, 57.33; H, 5.16; N, 4.78%; νmax (CHCl3) 3420 (w), 3500–3150 (br w), 1725 (s), 1600 (w) and 1270 (s) cm−1; δH (200 MHz, CDCl3) 2.53 (1H, dd, J = 18.0 Hz, J′ = 2.5 Hz, C(4)HH′), 3.06 (1H, dd, J = 18.0 Hz, J′ = 7.0 Hz, C(4)HH′), 3.65 (3H, s, CO2CH3), 3.94 (1H, d, J = 11.5 Hz, CHH′OH), 4.08 (1H, d, J = 11.5 Hz, CHH′OH), 4.72 (1H, br s, OH), 5.71 (1H, dd, J = 7.0 Hz, J′ = 2.5 Hz, C(3)H), 7.39–7.46 (2H, m, ArH), 7.54–7.61 (1H, m, ArH) and 7.91–7.99 (3H, m, ArH and NH); δC (50.3 MHz, CDCl3) 38.0 (C(4)H2), 52.7 (CO2CH3), 65.2, 71.8 and 72.4 (CH2OH, C(2) and C(3)H), 128.4, 128.8, 129.7 and 133.9 (ArC) and 164.8, 169.4 and 175.9 (CO); m/z (CI) 311 (M + NH4+, 30%) and 294 (M + H+, 100).(2R,3S)-3-Benzoyloxy-2-tert-butoxycarbonyloxymethyl-2-methoxycarbonylpyrrolidin-5-one 15b
To a solution of amido alcohol 15a (50 mg, 0.17 mmol) and pyridine (28 μl, 27 mg, 0.35 mmol) in DCM (3 ml) was added (BOC)2O (75 mg, 0.34 mmol). The mixture was stirred at room temperature for 5 h and solvent was then removed in vacuo. Purification by column chromatography (EtOAc–petrol 2 ∶ 3) gave carbonate 15b (62 mg, 92%) as colourless needles; mp 144.5–146 °C; Rf = 0.09 (EtOAc–petrol, 3 ∶ 7); [α]22D = +63.9 (c = 1.08 in CHCl3); found: C, 58.28; H, 5.67; N, 3.30; C19H23NO8 requires: C, 58.01; H, 5.89; N, 3.56%; νmax (CHCl3) 3430 (w), 1745 (s) and 1725 (s) cm−1; δH (200 MHz, CDCl3) 1.47 (9H, s, C(CH3)3), 2.56 (1H, dd, J = 17.5 Hz, J′ = 3.5 Hz, C(4)HH′), 2.99 (1H, dd, J = 17.5 Hz, J′ = 7.0 Hz, C(4)HH′), 3.68 (3H, s, CO2CH3), 4.29 (1H, d, J = 11.0 Hz, CHH′OBoc), 4.61 (1H, d, J = 11.0 Hz, CHH′OBoc), 5.70 (1H, dd, J = 7.0 Hz, J′ = 3.5 Hz, C(3)H), 6.70 (1H, br s, NH), 7.44 (2H, t, J = 7.5 Hz, ArH), 7.59 (1H, t, J = 7.5 Hz, ArH) and 7.94 (2H, d, J = 7.5 Hz, ArH); δC (50.3 MHz, CDCl3) 27.6 (C(CH3)3), 37.1 (C(4)H2), 53.1 (CO2CH3), 68.0, 69.1 and 71.6 (CH2OBoc, C(2) and C(3)H), 83.4 (C(CH3)3), 128.6, 129.6 and 133.8 (ArC), 152.7 (carbonate CO) and 164.8, 168.4 and 173.7 (amide and ester CO); m/z (CI) 411 (M + NH4+, 2%), 394 (M + H+, 2), 294 (100), 142 (50) and 105 (48).(2S)-1-tert-Butoxycarbonyl-2-tert-butoxycarbonyloxymethyl-2-methoxycarbonyl-2,5-dihydro-1H-pyrrol-5-one 16
To a solution of amido alcohol 15a (100 mg, 0.34 mmol), triethylamine (48 μl, 35 mg, 0.34 mmol) and DMAP (42 mg, 0.34 mmol) in DCM (5 ml) was added (Boc)2O (150 mg, 0.69 mmol). The mixture was stirred at room temperature for 5 h and solvent was then removed in vacuo. Purification by column chromatography (EtOAc–petrol, 1 ∶ 3 increasing polarity to EtOAc–petrol 2 ∶ 3) gave carbonate 15b (41 mg, 31%) as colourless needles and alkene 16 (73 mg, 58%) as a colourless oil; Rf = 0.34 (EtOAc–petrol, 3 ∶ 7); [α]22D = +34.7 (c = 2.39 in CHCl3); νmax (CHCl3) 2985 (m), 1790 (s), 1745 (s), 1370 (m) and 1330 (s) cm−1; δH (200 MHz, CDCl3) 1.41 and 1.51 (18H, 2 × s, 2 × C(CH3)3), 3.74 (3H, s, CO2CH3), 4.76 (1H, d, J = 12.0 Hz, CHH′OBoc), 4.87 (1H, d, J = 12.0 Hz, CHH′OBOC), 6.22 (1H, d, J = 6.5 Hz, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH′) and 7.00 (1H, d, J
= 6.5 Hz, CHCH′); δC (50.3 MHz, CDCl3) 27.3 and 27.7 (2 × C(CH3)3), 53.1 (CO2CH3), 63.5 and 72.3 (CH2OBoc and C(2)), 83.0 and 84.1 (2 ×
C(CH3)3), 128.8 and 145.8 (vinylic), 148.2 and 153.1 (2 × carbonate CO) and 167.2 and 168.6 (amide and ester CO); m/z (CI) 389 (M + NH4+, 10), 372 (M + H+, 12), 216 (100) and 142 (100).
CH′) and 7.00 (1H, d, J
= 6.5 Hz, CHCH′); δC (50.3 MHz, CDCl3) 27.3 and 27.7 (2 × C(CH3)3), 53.1 (CO2CH3), 63.5 and 72.3 (CH2OBoc and C(2)), 83.0 and 84.1 (2 ×
C(CH3)3), 128.8 and 145.8 (vinylic), 148.2 and 153.1 (2 × carbonate CO) and 167.2 and 168.6 (amide and ester CO); m/z (CI) 389 (M + NH4+, 10), 372 (M + H+, 12), 216 (100) and 142 (100).(2R,5R,6S,7S)-, (2R,5R,6S,7R)- and (2R,5R,6R,7S)-2-tert-Butyl-6-hydroxy-5-methoxycarbonyl-7-methyl-8-oxo-3-oxa-1-azabicyclo[3.3.0]octane 17a, 17b and 17c
To a deoxygenated solution of lactam 5b (617 mg, 2.3 mmol) in 10% AcOH in DCM (20 ml) under N2 was added, portionwise over 45 min, NaBH4 (265 mg, 7 mmol). The mixture was stirred at room temperature for 20 h, the solvent was evaporated in vacuo and the residue was partitioned between EtOAc (40 ml) and 5% NaHCO3 (aq) (40 ml). The aqueous layer was acidified with 2 M HCl and extracted with EtOAc (2 × 20 ml). The EtOAc extracts were washed with brine (30 ml), dried (MgSO4) and evaporated in vacuo to give unreacted starting material (5b) (181 mg, 29%). The organic layer was washed with brine (30 ml), dried (MgSO4) and evaporated in vacuo. Purification by careful multiple column chromatography (DCM–EtOAc, 1 ∶ 4 increasing polarity to DCM–EtOAc, 1 ∶ 1) gave the alcohols 17c (26 mg, 4%), 17a (197 mg, 32%) and 17b (100 mg, 16%) as colourless crystalline solids.Epimerisation of 17b with sodium methoxide
A solution of alcohol 17b (20 mg, 0.07 mmol) and NaOMe (40 mg, 0.74 mmol) in MeOH (3 ml) was stirred at room temperature for 15.75 h and then partitioned between EtOAc (10 ml) and brine (5 ml), the organic layer being dried (MgSO4) and evaporated in vacuo to give 20 mg of a 30 ∶ 1 mixture of alcohol 17a–alcohol 17b.Epimerisation of 17a with sodium methoxide
A solution of alcohol 17a (15 mg, 0.06 mmol) and NaOMe (30 mg, 0.55 mmol) in MeOH (2.5 ml) was stirred at 0 °C for 5.25 h and then partitioned between EtOAc (10 ml) and brine (5 ml), the organic layer being dried (MgSO4) and evaporated in vacuo to give 15 mg of alcohol 17a, no other diastereomers being seen in the NMR spectrum.(2R,5S)-2-tert-Butyl-5-methoxycarbonyl-7-methyl-8-oxo-3-oxa-1-azabicyclo[3.3.0]oct-6-ene 19
To a solution of triphenylphosphine (17 mg, 0.065 mmol), benzoic acid (8 mg, 0.066 mmol) and alcohol 17b (16 mg, 0.059 mmol) in THF (2.5 ml) under N2 was added dropwise a solution of diethyl azodicarboxylate (11 mg, 0.063 mmol) in THF (1.5 ml). The mixture was stirred at room temperature for 24 h and then partitioned between ether (5 ml) and 5% NaHCO3 (aq) (5 ml). The ether layer was washed with brine (5 ml), dried (MgSO4) and evaporated in vacuo. Purification by column chromatography (DCM increasing polarity to EtOAc–DCM, 1 ∶ 3) gave starting material 17b (6 mg, 38%) (after recrystallisation from CHCl3–hexane to remove triphenylphosphine oxide) and alkene 19 (5 mg, 33%) as a viscous oil; Rf = 0.13 (DCM); [α]22D = +210 (c = 0.22 in CHCl3); νmax (CHCl3) 2960 (m), 2870 (w), 1745 (s) and 1715 (s) cm−1; δH (200 MHz, CDCl3) 0.97 (9H, s, C(CH3)3), 1.92 (3H, d, J = 1.5 Hz, CHCCH3), 3.26 (1H, d, J
= 8.5 Hz, C(4)HH′), 3.76 (3H, s, CO2CH3), 4.69 (1H, s, tBuCH), 4.78 (1H, d, J
= 8.5 Hz, C(4)HH′) and 6.73 (1H, d, J
= 1.5 Hz, CHCCH3); δC (125.7 MHz, CDCl3) 11.04 (CHCCH3), 24.83 (C(CH3)3), 35.23 (C(CH3)3), 52.90 (CO2CH3), 71.11 (C(4)H2), 75.60 (C(5)),
96.85 (tBuCH), 138.6 and 139.3 (CHCCH3) and 169.9 and 178.3 (carbonyls); m/z (GCMS) 254 (M + H+, 100%) and 196 (15).(2R,3S)-3-Hydroxy-2-hydroxymethyl-2-methoxycarbonyl-4-methylpyrrolidin-5-one 21
To AcOH (2 ml) cooled in a water bath was added portionwise NaBH4 (30 mg, 0.79 mmol) and the mixture was stirred until H2 evolution ceased (about 10 min). A solution of keto alcohol 20 (80 mg, 0.40 mmol) in AcOH (0.5 ml) was then added dropwise and the mixture was stirred at room temperature for 2.75 h. Solvent was removed in vacuo and the residue was purified by column chromatography (EtOAc increasing polarity to MeOH–EtOAc, 1 ∶ 4) to give a 3 ∶ 2 mixture of the epimers of diol 21 (41 mg, 51%) as a colourless glass; Rf = 0.49 and 0.41 (MeOH–EtOAc, 1 ∶ 3); νmax (KBr) 3700–2600 (br s), 1730 (s) and 1695 (s) cm−1; δH (200 MHz, D2O) (major epimer) 1.00 (3H, d, J = 7.5 Hz, CHCH3), 2.40 (1H, quin, J = 7.5 Hz, C(4)H), 3.56 (1H, d, J = 12.0 Hz, CHH′OH), 3.61 (3H, s, CO2CH3), 3.89 (1H, d, J = 7.5 Hz, C(3)HOH) and 3.93 (1H, d, J = 12.0 Hz, CHH′OH); (minor epimer) 0.89 (3H, d, J = 7.5 Hz, CHCH3), 2.67 (1H, quin, J = 7.0 Hz, C(4)H), 3.55 (1H, d, J = 11.0 Hz, CHH′OH), 3.61 (3H, s, CO2CH3), 3.85 (1H, d, J = 11.0 Hz, CHH′OH) and 4.18 (1H, d, J = 6.0 Hz, C(3)HOH); m/z (CI) 221 (M + NH4+, 6%), 204 (M + H+, 100) and 144 (18).(2R,5R)-2-tert-Butyl-5-hydroxymethyl-6,8-dioxo-3-oxa-1-azabicyclo[3.3.0]octane 22a
To a solution of ester 4a (95 mg, 0.37 mmol) in THF (5 ml) at 0 °C was added a solution of DIBAL-H (0.94 M in hexane, 1.6 ml, 1.50 mmol). The reaction was stirred at 0 °C for 1 h, then quenched by the cautious addition of MeOH (1 ml) before partitioning between ether (20 ml) and 2 M HCl (20 ml). The aqueous layer was saturated with NaCl and re-extracted with EtOAc (20 ml), the combined organic extracts being washed with brine (30 ml), dried (MgSO4) and evaporated in vacuo to give crude alcohol 22a (72 mg, 85%) as a brown oil; δH (200 MHz, CDCl3) 0.96 (9H, s, C(CH3)3), 3.05 (1H, d, J = 21.5 Hz, C(7)HH′), 3.53 (1H, d, J = 21.5 Hz, C(7)HH′), 3.56 (1H, d, J = 9.0 Hz, C(4)HH′), 3.78 (1H, d, J = 9.0 Hz, C(4)HH′), 3.82 (1H, d, J = 11.0 Hz, CHH′OH), 3.98 (1H, d, J = 11.0 Hz, CHH′OH) and 4.98 (1H, s, tBuCH); δC (50.3 MHz, CDCl3) 25.3 (C(CH3)3), 35.0 (C(CH3)3), 46.9 (C(7)H2), 63.7 and 67.2 (CH2OH and C(4)H2), 79.7 (C(5)), 98.5 (tBuCH), 174.5 (amide CO) and 206.1 (ketone CO).(2R,5R,6R)-2-tert-Butyl-6-hydroxy-5-hydroxymethyl-8-oxo-3-oxa-1-azabicyclo[3.3.0]octane 23
NaBH4 (63 mg, 1.67 mmol) was added to AcOH (1.5 ml) and stirred at room temperature for 15 min. After H2 evolution had ceased a solution of crude alcohol 22a (38 mg, 0.17 mmol) in acetone (1.5 ml) was added and the mixture was stirred at room temperature for 2.5 h. Solvent was removed in vacuo and the residue was partitioned between EtOAc (10 ml) and 5% aq NaHCO3 (5 ml). The aqueous layer was re-extracted with EtOAc (10 ml) and the combined organic layers were washed with brine (10 ml), dried (MgSO4) and evaporated in vacuo. Purification by column chromatography (EtOAc–petrol, 3 ∶ 1 increasing polarity to EtOAc) gave diol 23 (27 mg, 70%) as colourless crystals; mp 183–184.5 °C (decomp.); Rf = 0.17 (EtOAc); [α]21D = +77.3 (c = 0.33 in MeOH); found: C, 57.3; H, 8.6; N, 5.8; C11H19NO4 requires: C, 57.6; H, 8.4; N, 6.1%; νmax (KBr) 3410 (m), 3365 (m), 2970 (m), 1695 (s) and 1080 (m) cm−1; δH (200 MHz, CD3OD) 0.93 (9H, s, C(CH3)3), 2.21 (1H, d, J = 16.5 Hz, C(7)HH′), 3.28 (1H, dd, J = 16.5 Hz, J′ = 5.5 Hz, C(7)HH′), 3.60 (1H, d, J = 11.0 Hz, CHH′OH), 3.70 (1H, d, J = 11.0 Hz, CHH′OH), 3.78 (1H, d, J = 9.0 Hz, C(4)HH′), 4.03 (1H, d, J = 9.0 Hz, C(4)HH′), 4.41 (1H, d, J = 5.5 Hz, C(6)H) and 4.73 (1H, s, tBuCH); δC (50.3 MHz, CD3OD) 26.2 (C(CH3)3), 36.5 (C(CH3)3), 46.0 (C(7)H2), 64.9, 67.9 and 70.6 (CH2OH, C(4)H2 and C(6)H), 77.2 (C(5)), 97.3 (tBuCH) and 180.5 (amide CO); m/z (CI) 230 (M + H+, 100%) and 172 (33).(2R,5R)-2-tert-Butyl-6,8-dioxo-5-(2′-methylpropanoyl)-3-oxa-1-azabicyclo[3.3.0]octane 24a
To a suspension of Li (0.5% Na, 30% dispersion in mineral oil) (1.00 g, 43.2 mmol) in petrol (15 ml) was added isopropyl chloride (0.3 ml, 0.26 g, 3.3 mmol). The flask was placed in a sonicator for 5 minutes and isopropyl chloride (1.5 ml, 1.29 g, 16.4 mmol) was then added over 1 h so as to keep the mixture at a gentle reflux. After stirring for a further 1 h the reaction was cooled to −78 °C and a solution of ester 5a (0.89 g, 3.49 mmol) in THF (15 ml) was added dropwise over 10 min. After 3 h at −78 °C the reaction was quenched by the cautious addition of water. After warming to room temperature the mixture was partitioned between ether (50 ml) and water (50 ml). The aqueous layer was acidified with 2 M HCl and extracted with ether (2 × 30 ml), the ether extract being washed with brine (40 ml), dried (MgSO4) and evaporated in vacuo to give an inseparable 3 ∶ 1 mixture of ketone 24a and alcohol 25a (0.89 g) as a pale yellow crystalline solid; mp 191–192.5 °C; Rf = 0.32 (ketone), 0.36 (alcohol) (AcOH–EtOAc–DCM, 1 ∶ 20 ∶ 80); νmax (KBr) 3600–3300 (br w), 3590 (w), 2980 (m), 2800–2100 (br w), 1730 (s), 1650 (s), 1575 (s), 1480 (s), 1315 (s) and 1295 (s) cm−1; δH (500 MHz, CD3OD) 0.87 (9H, s, C(CH3)3), 1.087 (3H, d, J = 7.1 Hz, CH(CH3)(CH3)′), 1.09 (3H, d, J = 6.5 Hz, CH(CH3)(CH3)′), 3.33 (1H, sept, J = 6.8 Hz, CH(CH3)2), 3.42 (1H, d, J = 8.5 Hz, C(4)HH′), 4.58 (1H, s, tBuCH) and 4.84 (1H, d, J = 8.5 Hz, C(4)HH′); δC (125.7 MHz, CD3OD) 19.47 and 21.78 (CH(CH3)2), 25.80 (C(CH3)3), 35.53 (CH(CH3)2), 35.85 (C(CH3)3), 68.95 (C(4)H2), 76.68 (C(5)), 96.34 (CDCOH, JCD
= 27 Hz), 99.03 (tBuCH), 178.8 (amide CO), 183.8 (CDCOD) and 209.1 (ketone CO); m/z (CI) 285 (M + NH4+, 3%), 268 (M + H+, 100), 210 (24) and 197 (26).Data for 25a are given below.
(2R,5R)-2-tert-Butyl-6-hydroxy-7-methyl-8-oxo-5-(2′-methylpropanoyl)-3-oxa-1-azabicyclo[3.3.0]oct-6-ene 24b
To a suspension of Li (0.5% Na, 30% dispersion in mineral oil) (230 mg, 9.94 mmol) in petrol (10 ml) was added isopropyl chloride (100 μl, 86 mg, 1.1 mmol). The flask was placed in a sonicator for 5 minutes and isopropyl chloride (300 μl, 238 mg, 3.3 mmol) was then added in 3 portions over 30 min. After stirring for a further 1.5 h the reaction was cooled to −78 °C and a solution of ester 5b (237 mg, 0.88 mmol) in THF (10 ml) was added dropwise over 10 min. After 2 h at −78 °C the reaction was quenched by the cautious addition of water. After warming to room temperature the mixture was partitioned between ether (40 ml) and water (40 ml). The aqueous layer was acidified with 2 M HCl and extracted with ether (2 × 25 ml), the ether extract being washed with brine (40 ml), dried (MgSO4) and evaporated in vacuo. Recrystallisation from EtOAc–CHCl3–petrol gave ketone 24b (62 mg, 25%) as colourless crystals. A second crop (82 mg, 33%) was obtained containing alcohol 25b as a minor component (<4%). The residue was a 3 ∶ 2 mixture of ketone 24b–alcohol 25b [65 mg, 16% ketone (24b), 10% alcohol (25b)]; mp 188–190.5 °C; [α]23D = +286 (c = 0.94 in MeOH); νmax (KBr) 3440 (br s), 2980 (s), 2680 (br m), 1730 (s), 1655 (s), 1605 (s), 1325 (s) and 1175 (s) cm−1; δH (200 MHz, CD3OD) 0.87 (9H, s, C(CH3)3), 1.04 (3H, d, J = 7.0 Hz, CH(CH3)(CH3)′), 1.06 (3H, d, J = 7.0 Hz, CH(CH3)(CH3)′), 1.69 (3H, s, CH3CCOH), 3.24 (1H, sept, J
= 7.0 Hz, CH(CH3)2), 3.33 (1H, d, J
= 8.5 Hz, C(4)HH′), 4.83 (1H, s, tBuCH) and 4.95 (1H, d, J
= 8.5 Hz, C(4)HH′); δC (50.3 MHz, CDCl3) 4.8 (CH3CCOH), 18.0 and 20.2 (CH(CH3)2), 24.3 (C(CH3)3), 33.6 and 34.3 (CH(CH3)2 and C(CH3)3), 67.4 (C(4)H2), 79.0 (C(5)), 97.4 (tBuCH), 103.8 (CH3CCOH), 170.5 (amide CO), 183.1 (CH3C =
COH) and 208.9 (ketone CO); m/z (CI) 299 (M + NH4+,
8%) and 282 (M + H+, 100).(2R,5S,1′R)-2-tert-Butyl-6,8-dioxo-5-(1′-hydroxy-2′-methylpropyl)-3-oxa-1-azabicyclo[3.3.0]octane 25a
Ammonia (12 ml) was condensed in a flask fitted with a dry ice condenser at −78 °C and Li metal (0.35 g, 50.4 mmol) was then added. After 15 min at −78 °C a solution of ketone 24a (contaminated with 25% alcohol 25a) (100 mg, 0.37 mmol) in THF (4 ml) was added to the bronze solution and followed after a further 5 min by ethanol (1 ml). The mixture was allowed to warm to −33 °C over 10 min and further ethanol (1 ml) was added. After 30 min at −33 °C the reaction was quenched by the addition of excess ethanol (5 ml) and the mixture was allowed to warm to room temperature. The reaction mixture was partitioned between ether (15 ml) and water (15 ml), the ether layer being extracted with sat. NaHCO3 (aq) (2 × 15 ml). The combined aqueous layers were acidified with conc. HCl and extracted with ether (2 × 20 ml), the ether layers being washed with brine (25 ml), dried (MgSO4) and evaporated in vacuo to give alcohol 25a (74 mg, 70% from ester 5a) as a pale pink crystalline solid; mp 168–170 °C; Rf = 0.36 (AcOH–EtOAc–DCM, 1 ∶ 20 ∶ 80); [α]23D = +98.2 (c = 1.14 in CHCl3); νmax (KBr) 3600–2200 (br m), 2960 (s), 1680 (s), 1650 (s), 1610 (s) and 1290 (s) cm−1; δH (200 MHz, CDCl3) 0.84 (3H, d, J = 7.0 Hz, CH(CH3)(CH3)′), 0.99 (9H, s, C(CH3)3), 1.07 (3H, d, J = 7.0 Hz, CH(CH3)(CH3)′), 2.14 (1H, d sept, Jsept = 7.0 Hz, Jd = 1.5 Hz, CH(CH3)2), 2.56 (1H, br s, OH), 3.22 (1H, d, J = 22.0 Hz, C(7)HH′), 3.46 (1H, d, J = 9.0 Hz, C(4)HH′), 3.54 (1H, d, J = 22.0 Hz, C(7)HH′), 3.77 (1H, br s, CHOH(iPr)), 4.51 (1H, d, J = 9.0 Hz, C(4)HH′) and 4.97 (tBuCH); δC (50.3 MHz, CDCl3) 16.2 and 21.7 (CH(CH3)2), 25.6 (C(CH3)3), 27.9 (CH(CH3)2), 34.9 (C(CH3)2), 46.5 (C(7)H2), 69.1, 77.0 and 79.3 (C(4)H2, C(5) and CHOH(iPr)), 99.7 (tBuCH), 174.2 (amide CO) and 208.0 (ketone CO); m/z (CI) 270 (M + H+, 63%), 198 (59) and 140 (100).(2R,5R,1′R)-2-tert-Butyl-6-hydroxy-7-methyl-8-oxo-5-(1′-hydroxy-2′-methylpropyl)-3-oxa-1-azabicyclo[3.3.0]octane 25b
COH), 2.23 (1H, sept, J
= 7.0 Hz, CH(CH3)2), 3.37 (1H, d, J
= 8.5 Hz, C(4)HH′), 3.62 (1H, br s, CHOH), 4.06 (1H, br s, CHOH(iPr)), 4.37 (1H, d, J
= 8.5 Hz, C(4)HH′), 4.57 (1H, s, tBuCH) and 8.32 (1H, br s, CH3CCOH); δC (50.3 MHz, CDCl3)
5.5 (CH3CCOH), 15.1 and 21.4 (CH(CH3)2), 25.3 (C(CH3)3), 27.6 (CH(CH3)2), 34.6 (C(CH3)3), 71.0, 72.6 and 78.0 (C(4)H2, C(5) and CHOH(iPr)), 97.7 (tBuCH), 103.9 (CH3CCOH), 172.7 (amide CO) and 182.2 (CH3CCOH); m/z (CI) 317 (M + O + NH4+, 3%), 300 (M + O + H+, 10), 284 (M + H+, 100).(2R,5S,1′S)-2-tert-Butyl-6,8-dioxo-5-(1′-hydroxy-2′-methylpropyl)-3-oxa-1-azabicyclo[3.3.0]octane 26
(2R,5S,6S,1′S)-2-tert-Butyl-6-hydroxy-8-oxo-5-(1′-hydroxy-2′-methylpropyl)-3-oxa-1-azabicyclo[3.3.0]octane 27
A solution of alcohol 26 (24 mg, 0.09 mmol) and PtO2 (10 mg) in EtOAc (3 ml) was stirred at room temperature under 4–5 atm pressure of H2 (Fischer bottle) for 16.5 h. The crude reaction mixture was then filtered through Celite® to remove PtO2 and evaporated in vacuo. Purification by column chromatography (EtOAc–petrol, 3 ∶ 7 increasing polarity to EtOAc–petrol, 1 ∶ 1) gave unreacted starting material 26 (18 mg, 75%) and diol 27 (6 mg, 25%) as an amorphous solid; mp 144–146 °C (decomp.); Rf = 0.18 (EtOAc–petrol, 1 ∶ 1); [α]23D = +95.3 (c = 0.30 in CHCl3); νmax (CHCl3) 3555 (m), 3315 (br m), 1670 (s) and 1350 (m) cm−1; δH (200 MHz, CDCl3) 1.00 (9H, s, C(CH3)3, 1.07 (6H, d, J = 6.5 Hz, CH(CH3)2), 2.25 (1H, oct, J = 6.5 Hz, CH(CH3)2), 2.67 (1H, br d, J = 5.0 Hz, CHOH(iPr)), 2.85 (1H, dd, J = 16.5 Hz, J′ = 8.5 Hz, C(7)HH′), 3.04 (1H, dd, J = 16.5 Hz, J′ = 9.5 Hz, C(7)HH′), 3.32 (1H, d, J = 9.5 Hz, C(4)HH′), 3.41 (1H, br d, J = 10.5 Hz, C(6)HOH), 3.85 (1H, dd, J = 6.5 Hz, J′ = 5.0 Hz, CHOH(iPr)), 4.38 (1H, d, J = 9.5 Hz, C(4)HH′), 4.41 (1H, q, J = 9.0 Hz, C(6)HOH) and 4.78 (1H, s, tBuCH); δC (50.3 MHz, CDCl3) 18.9 and 23.1 (CH(CH3)2), 26.0 (C(CH3)3, 30.2 (CH(CH3)2), 35.1 (C(CH3)3, 46.3 (C(7)H2), 73.6, 74.4, 76.7 and 79.2 (C(4)H2, C(5), C(6)H and CHOH(iPr)), 97.0 (tBuCH) and 177.4 (amide CO); m/z (CI) 272 (M + H+, 100%) and 214 (15).(2R,5R,1′S)-2-tert-Butyl-6-hydroxy-7-methyl-8-oxo-5-(1′-hydroxy-2′-methylpropyl)-3-oxa-1-azabicyclo[3.3.0]oct-6-ene 28
To a solution of ketone 24b (50 mg, 0.18 mmol) in degassed EtOH (5 ml) under N2 was added NaOMe (10 mg, 0.19 mmol). After stirring at room temperature for 10 min NaBH4 (14 mg, 0.37 mmol) was added and the mixture was stirred for a further 15 h. After partitioning between ether (20 ml) and 2 M HCl (20 ml) the aqueous layer was washed with ether (15 ml), the combined organic extracts being washed with brine (25 ml), dried (MgSO4) and evaporated in vacuo. The crude material was triturated with cold ether, and the ether washings were concentrated in vacuo and purification of this material by column chromatography (EtOAc–petrol, 3 ∶ 7) gave further alcohol 28 (2 mg, 4%) as a white powder; mp 173–174.5 °C; Rf = 0.19 (EtOAc–petrol, 3 ∶ 7); [α]25D = +94.0 (c = 0.72 in MeOH); νmax (KBr) 3565 (m), 3435 (br m), 2960 (s), 1655 (m) and 1610 (s) cm−1; δH (200 MHz, CD3OD) (enol tautomer) 1.00 (9H, s, C(CH3)3, 1.03 (6H, d, J = 6.5 Hz, CH(CH3)2), 1.62 (3H, s, CH3CCOH), 2.12 (1H, oct, J
= 6.5 Hz, CH(CH3)2), 3.44 (1H, d, J
= 9.0 Hz, C(4)HH′), 3.62 (1H, d, J
= 6.5 Hz, CHOH(iPr)), 4.27 (1H, d, J
= 9.0 Hz, C(4)HH′) and 4.45 (1H, s, tBuCH); (keto tautomer) 0.95 (3H, d, J
= 6.5 Hz, CH(CH3)(CH3)′), 1.88 (1H, oct, J
= 6.5 Hz, CH(CH3)2), 3.57 (1H, d, J
= 6.5
Hz, CHOH(iPr)), 3.63 (1H, m, C(4)HH′) and 4.14 (1H, d, J
= 9.5 Hz, C(4)HH′); δC (50.3 MHz, CD3OD) (enol tautomer) 6.5 (CH3CCOH), 20.8 and 22.7 (CH(CH3)2), 26.6 (C(CH3)3, 31.7 (CH(CH3)2), 36.1 (C(CH3)3, 71.6, 76.9 and 77.5 (C(4)H2, C(5) and CHOH(iPr)), 98.7 (tBuCH), 104.5 (CH3CCOH), 174.2 (amide CO) and 184.2 (CH3CCOH); (keto tautomer) 7.1 (C(7)CH3), 19.4 and 22.5 (CH(CH3)2), 26.3 (C(CH3)3, 32.0 (CH(CH3)2) and 69.8 and 78.0 (C(4)H2 and CHOH(iPr)); m/z
(CI) 284 (M + H+, 100%), 226 (M + H+
−
tBuH, 54) and 212 (46); exact mass 284.1859, C15H25NO4 (MH+) requires 284.1862.(2R,5S,1′S)-2-tert-Butyl-7-hydroxy-7-methyl-6,8-dioxo-5-(1′-hydroxy-2′-methylpropyl)-3-oxa-1-azabicyclo[3.3.0]octane 29
Attempted recrystallisation of crude alcohol 25b (38 mg, 0.14 mmol), formed by the NaBH4 reduction of ketone 24b, from hot CHCl3 gave decomposition. Purification of the residue by column chromatography (EtOAc–petrol, 1 ∶ 4) gave, apart from starting material, the autoxidation product 29 (3 mg, 7%) as a colourless oil; Rf = 0.55 (EtOAc–petrol, 3 ∶ 7); [α]23D = +71 (c = 0.12 in CHCl3); νmax (film) 3600–3100 (br m), 2960 (m), 1770 (m) and 1705 (s) cm−1; δH (200 MHz, CDCl3) 1.04 (3H, d, J = 7.0 Hz, CH(CH3)(CH3)′), 1.05 (9H, s, C(CH3)3, 1.13 (3H, d, J = 7.0 Hz, CH(CH3)(CH3)′), 1.36 (3H, s, C(7)CH3), 1.94 (1H, oct, J = 7.0 Hz, CH(CH3)2), 2.96 (1H, br s, CHOH), 3.55 (1H, br d, J = 7.0 Hz, CHOH(iPr)), 3.60 (1H, d, J = 9.5 Hz, C(4)HH′), 4.22 (1H, d, J = 9.5 Hz, C(4)HH′), 4.95 (1H, s, tBuCH) and 5.23 (1H, br s, OH); δC (125.7 MHz, CDCl3) 16.61, 16.71 and 22.26 (CH(CH3)2 and C(7)CH3), 25.87 (C(CH3)3, 28.04 (CH(CH3)2), 34.69 (C(CH3)3, 68.98, 76.58 and 86.94 (C(4)H2, C(5), C(7) and CHOH), 100.9 (tBuCH), 177.6 (amide CO) and 209.7 (ketone CO); m/z (CI) 317 (M + NH4+, 18%), 300 (M + H+, 91), 245 (77) and 228 (100); m/z (electrospray) 317 (M + NH4+, 6%) and 300 (M + H+, 100).(2R,5S,1′R)-2-tert-Butyl-7-methyl-6,8-dioxo-7-peroxy-5-(1′-hydroxy-2′-methylpropyl)-3-oxa-1-azabicyclo[3.3.0]octane 30
A solution of alcohol 25b (12 mg, 0.04 mmol) in CDCl3 (0.5 ml) was left at room temperature for 20 days. Purification by column chromatography (EtOAc–petrol, 1 ∶ 4) gave starting material (6 mg, 50%) and peroxide 30 (4 mg, 30%) as a yellow oil (positive peroxide test with starch/KI paper); Rf = 0.30 (EtOAc–petrol, 3 ∶ 7); [α]23D = +48 (c = 0.10 in CHCl3); νmax (film) 3600–3100 (br m), 2965 (m), 1770 (m) and 1715 (s) cm−1; δH (200 MHz, CDCl3) 0.86 (3H, d, J = 7.0 Hz, CH(CH3)(CH3)′), 1.02 (9H, s, C(CH3)3, 1.08 (3H, d, J = 7.0 Hz, CH(CH3)(CH3)′), 1.47 (3H, s, C(7)CH3), 1.93 (1H, d sept, Jsept = 7.0 Hz, Jd = 2.0 Hz, CH(CH3)2), 2.50 (1H, br s, CHOH), 3.60 (1H, d, J = 8.5 Hz, C(4)HH′), 3.75 (1H, br s, CHOH(iPr)), 4.60 (1H, d, J = 8.5 Hz, C(4)HH′) and 5.02 (1H, s, tBuCH); δC (125.7 MHz, CDCl3) 15.07, 19.06 and 21.20 (CH(CH3)2 and C(7)CH3), 25.56 (C(CH3)3, 29.68 (CH(CH3)2), 35.32 (C(CH3)3, 69.08, 75.60, 77.40 and 78.91 (C(4)H2, C(5), C(7) and CHOH), 97.65 (tBuCH), 175.0 (amide CO) and 204.2 (ketone CO); m/z (CI) 317 (M − O + NH4+, 9%), 300 (M − O +H+, 28), 298 (M + H+ − H2O, 11) and 200 (100); m/z (electrospray) 316 (M + H+, 75%), 270 (72) and 200 (100).(3S,4S,9S,10S)-4,10-Dibenzoyloxy-2,8-dihydroxy-3,9-dimethoxycarbonyl-6,12-dioxo-1,7-diazatricyclo[7.3.0.03,7]dodecane 32
To a solution of (COCl)2 (35 μl, 51 mg, 0.40 mmol) in DCM (3 ml) under N2 at −78 °C was added DMSO (45 μl, 50 mg, 0.63 mmol). The mixture was stirred at −78 °C for 15 min and then a solution of alcohol 15a (100 mg, 0.34 mmol) in DCM (1.5 ml) was added dropwise. After a further 15 min at −78 °C triethylamine (140 μl, 102 mg, 1.0 mmol) was added and the mixture was allowed to warm to room temperature over 10 min. After stirring at room temperature for 3 h the mixture was partitioned between DCM (10 ml) and H2O (10 ml). The aqueous layer was re-extracted with DCM (10 ml) and the combined organic layers were dried (MgSO4) and evaporated in vacuo. Purification by column chromatography (EtOAc–petrol, 1 ∶ 1 increasing polarity to EtOAc–petrol, 3 ∶ 1) gave dimer 32 (42 mg, 42%) as an off-white solid; mp 185.5–188 °C (decomp.); Rf = 0.20 (EtOAc–petrol, 1 ∶ 1); [α]22D = +50.3 (c = 1.49 in CHCl3); νmax (CHCl3) 3560 (w), 3440 (br w), 2960 (w), 1730 (s) and 1605 (w) cm−1; δH (200 MHz, CDCl3) 2.95 (4H, d, J = 9.5 Hz, 2 × CH2CH), 3.54 (6H, s, 2 × CO2CH3), 5.55 (2H, d, exchangeable, J = 8.0 Hz, 2 × CHOH), 6.03 (2H, t, J = 9.5 Hz, 2 × CH2CH), 6.36 (2H, d, J = 8.0 Hz, 2 × CHOH), 7.41–7.48 (4H, m, ArH), 7.57–7.64 (2H, m, ArH) and 7.94 (4H, d, J = 7.0 Hz, ArH); δC (50.3 MHz, CDCl3) 35.4 (CH2CH), 53.7 (CO2CH3), 70.6 and 73.2 (CH2CH and CHOH), 72.3 (NCCO2CH3), 128.3 (ArC), 128.8, 129.7 and 134.1 (ArC) and 165.9, 168.6 and 172.7 (CO); m/z (CI) 583 (M + H+, <1%), 309 (M/2 + NH4+, 100), 292 (M/2 + H+, 93), 142 (76) and 105 (85); exact mass 292.0828, C14H14NO6 (M/2 + H+) requires 292.0821.(2R,3S,E)-3-Benzoyloxy-2-methoxycarbonyl-2-(2′ethoxycarbonylvinyl)pyrrolidin-5-one 33
To a solution of dimer 32 (72 mg, 0.12 mmol) in toluene (10 ml) was added ethoxycarbonylmethylenetriphenylphosphorane (90 mg, 0.26 mmol). The mixture was heated at reflux for 4 h, cooled to room temperature and partitioned between ether (25 ml) and NH4Cl (aq) (25 ml). The organic layer was washed with brine (20 ml), dried (MgSO4) and evaporated in vacuo. Purification by column chromatography gave a 2 ∶ 9 mixture of (Z)-alkene–(E)-alkene 33 (22 mg, 26%) as a pale yellow oil and pure (E)-alkene 33 (28 mg, 33%) as a pale yellow glass; Rf = 0.42 (EtOAc–petrol, 1 ∶ 1); [α]25D = +54.7 (c = 1.35 in CHCl3); νmax (CHCl3) 3430 (w), 1725 (s) and 1270 (s) cm−1; δH (200 MHz, CDCl3) 1.30 (3H, t, J = 7.0 Hz, CO2CH2CH3), 2.50 (1H, d, J = 17.5 Hz, C(4)HH′), 2.87 (1H, dd, J = 17.5 Hz, J′ = 5.5 Hz, C(4)HH′), 3.68 (3H, s, CO2CH3), 4.22 (2H, q, J = 7.0 Hz, CO2CH2CH3), 5.73 (1H, d, J = 5.5 Hz, C(3)H), 6.21 (1H, d, J = 15.5 Hz, CHCH′), 7.26 (1H, d, J
= 15.5 Hz, CHCH′), 7.41–7.48 (3H, m, ArH and NH), 7.56–7.64 (1H, m, ArH) and 7.95 (2H, d, J
= 7.0 Hz, ArH); δC (50.3 MHz, CDCl3) 14.1 (CO2CH2CH3), 36.3 (C(4)H2), 53.4
(CO2CH3), 61.2 (CO2CH2CH3), 71.1 and 74.1 (C(2) and C(3)H), 123.8 and 142.5 (vinylic), 128.5, 128.6, 129.7 and 133.9 (ArC) and 164.7, 165.0, 167.8 and 174.3 (CO); m/z (CI) 379 (M + NH4+, 100%), 362 (M + H+, 46), 257 (94), 240 (100), 208 (89) and 105 (70); exact mass 362.1234, C18H20NO7 (MH+) requires 362.1240.(Z)-isomer: Rf
= 0.38 (EtOAc–petrol, 1 ∶ 1); δH (200 MHz, CDCl3) 1.28 (3H, t, J
= 7.0 Hz, CO2CH2CH3), 2.71 (1H, dd, J
= 17.0 Hz, J′
= 6.0 Hz, C(4)HH′), 2.95 (1H, dd, J
= 17.0 Hz, J′
= 7.5 Hz, C(4)HH′), 3.65 (3H, s, CO2CH3), 4.20 (2H, q, J
= 7.0 Hz, CO2CH2CH3), 5.79 (1H, dd, J
= 7.5 Hz, J′
= 6.0 Hz, C(3)H), 6.08 (1H, d, J
= 11.5 Hz, CHCH′), 6.68 (1H, d, J
= 11.5 Hz, CHCH′), 7.4–8.0 (6H, ArH and NH).
Acknowledgements
We thank EPSRC and AstraZeneca for funding of a studentship to MDA, and we wish to gratefully acknowledge the use of the EPSRC Chemical Database Service at Daresbury24 and the EPSRC National Mass Spectrometry Service Centre at Swansea.References
- M. G. Moloney, Nat. Prod. Rep., 1999, 16, 485 RSC.
- S. Omura, T. Fujimoto, K. Otoguro, K. Matsuzaki, R. Moriguchi, H. Tanaka and Y. Sasaki, J. Antibiot., 1991, 44, 113 Search PubMed.
- E. J. Corey, W. Li and T. Nagamistu, Angew. Chem., Int. Ed., 1998, 37, 1676 CrossRef CAS.
- S. N. Crane and E. J. Corey, Org. Lett., 2001, 3, 1395 CrossRef CAS.
- E. J. Corey, W.-D. Z. Li, T. Nagamitsu and G. Fenteany, Tetrahedron Lett., 1999, 55, 3305 CrossRef CAS.
- J. H. Bailey, D. Cherry, J. Dyer, M. G. Moloney, M. J. Bamford, S. Keeling and R. B. Lamont, J. Chem. Soc., Perkin Trans. 1, 2000, 2783 RSC.
- J. Dyer, S. Keeling, A. King and M. G. Moloney, J. Chem. Soc., Perkin Trans. 1, 2000, 2793 RSC.
- J. H. Bailey, A. T. J. Byfield, P. J. Davis, A. C. Foster, M. Leech, M. G. Moloney, M. Muller and C. K. Prout, J. Chem Soc, Perkin Trans. 1, 2000, 1977 RSC.
- C. E. M. Davis, T. D. Heightman, S. A. Hermitage, M. G. Moloney and G. A. Woods, Tetrahedron Lett., 1998, 39, 1025 CrossRef.
- D. Seebach and J. D. Aebi, Tetrahedron Lett., 1984, 25, 2545 CrossRef CAS.
- M. D. Andrews, A. G. Brewster, K. M. Crapnell, A. J. Ibbett, T. Jones, M. G. Moloney, C. K. Prout and D. Watkin, J. Chem. Soc., Perkin Trans. 1, 1998, 223 RSC.
- M. D. Andrews, A. G. Brewster and M. G. Moloney, Synlett, 1996, 612 CrossRef CAS.
- M. D. Andrews, A. G. Brewster, J. Chuhan, A. J. Ibbett, M. G. Moloney, K. Prout and D. Watkin, Synthesis, 1997, 305 CrossRef CAS.
- M. D. Andrews, A. G. Brewster, M. G. Moloney and K. L. Owen, J. Chem. Soc., Perkin Trans. 1, 1996, 227 RSC.
- N. Galeotti, J. Poncet, L. Chiche and P. Jouin, J. Org. Chem., 1993, 58, 5370 CrossRef CAS.
- E. J. Corey and G. A. Reichard, J. Am. Chem. Soc., 1992, 114, 10677 CrossRef CAS.
- M. D. Andrews, A. G. Brewster, J. Chuhan, M. G. Moloney and C. K. Prout, unpublished results.
- D. L. Hughes, Org. React. (N. Y.), 1992, 42, 335 Search PubMed.
- R. W. Armstrong and J. A. DeMattei, Tetrahedron Lett., 1991, 32, 5749 CrossRef CAS.
- I. Kikkawa and T. Yorifuji, Synthesis, 1980, 877 CrossRef CAS.
- D. Bryce-Smith and E. E. Turner, J. Chem. Soc., 1953, 861 RSC.
- B. J. Wakefield, Organolithium Methods, Academic, London, 1988 Search PubMed.
- J. W. Huffman, in Comprehensive Organic Synthesis; Pergamon, Oxford, 1991, p. 107 Search PubMed.
- D. A. Fletcher, R. F. McMeeking and D. Parkin, J. Chem. Inf. Comput. Sci., 1996, 36, 746 CrossRef.
Footnote |
| † This compound exists as the enol tautomer, but is drawn in the keto form for convenience. |
| This journal is © The Royal Society of Chemistry 2002 |